
IMG_1530.jpg
Morphology of IE
Reading File
analyse_media
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a complete breakdown of the Morphology of Infective Endocarditis (IE), compiled from your notes and cross-referenced with Robbins Pathology:
Morphology of Infective Endocarditis (IE)
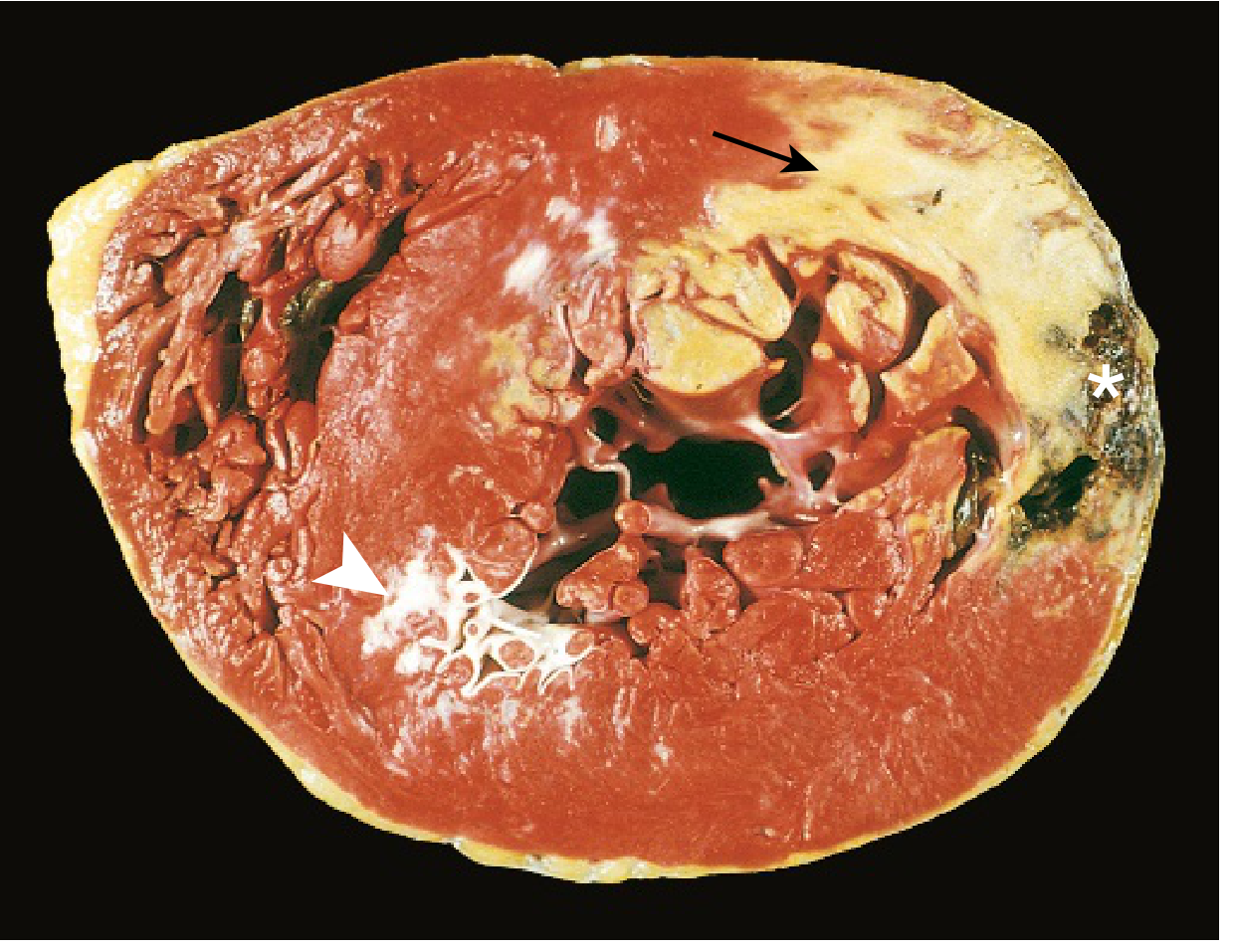
Gross Features
| Feature | Detail |
|---|---|
| Vegetation character | Friable, bulky, and potentially destructive |
| Composition | Fibrin, inflammatory cells, and organisms |
| Valves most affected | Aortic and mitral (left-sided, high pressure) |
| Right-sided disease | Seen in IV drug users (tricuspid valve most common) |
| Number | Single or multiple |
| Extension | Can erode into underlying myocardium → ring abscess |
| Emboli | Vegetation fragments embolize → lodge at distant sites → septic infarcts / mycotic aneurysms |
Ring abscess = when vegetation erodes into the myocardium at the valve annulus. Most feared complication of acute IE (especially S. aureus).
Comparing the 4 forms of endocarditis (vegetations):
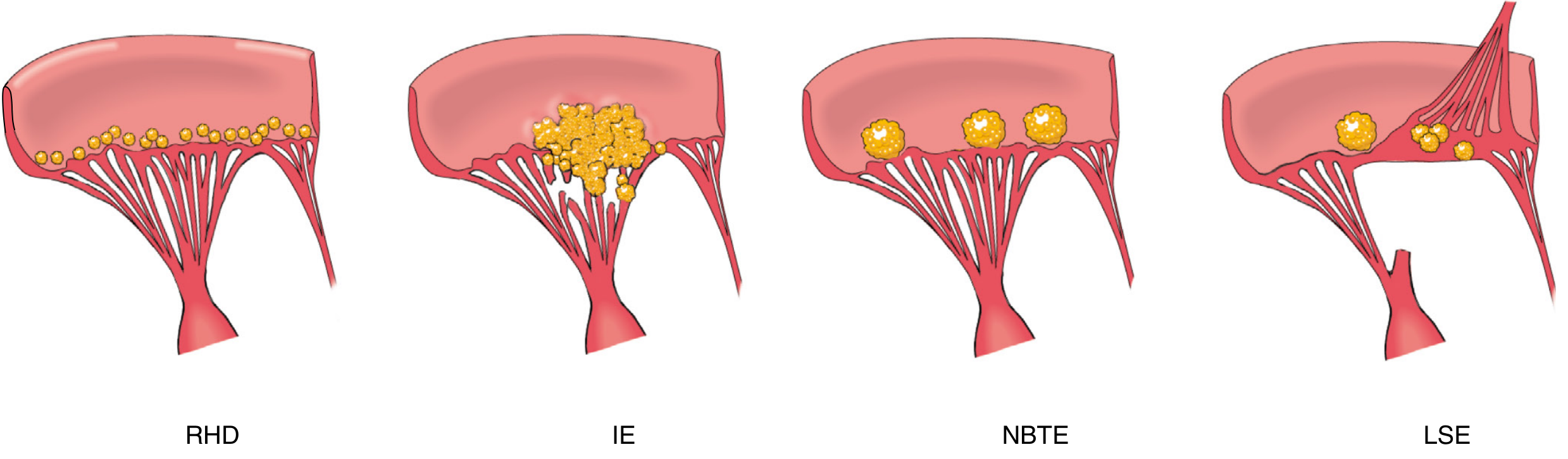
Microscopy
| IE Type | Microscopic Finding |
|---|---|
| Both | Vegetation = fibrin + inflammatory cells + organisms |
| Subacute IE | Granulation tissue at the base of vegetation (= healing / organization) |
| Chronic (late) | Fibrosis, calcification, chronic inflammatory infiltrate at base |
| Acute IE | More destructive - less evidence of healing/granulation at base |
The key microscopic distinction:
- Acute IE - minimal healing; rapid destruction
- Subacute IE - granulation tissue at base indicates attempted repair (organism has lower virulence, e.g., Strep viridans)
Gross Pathology Images (Robbins)
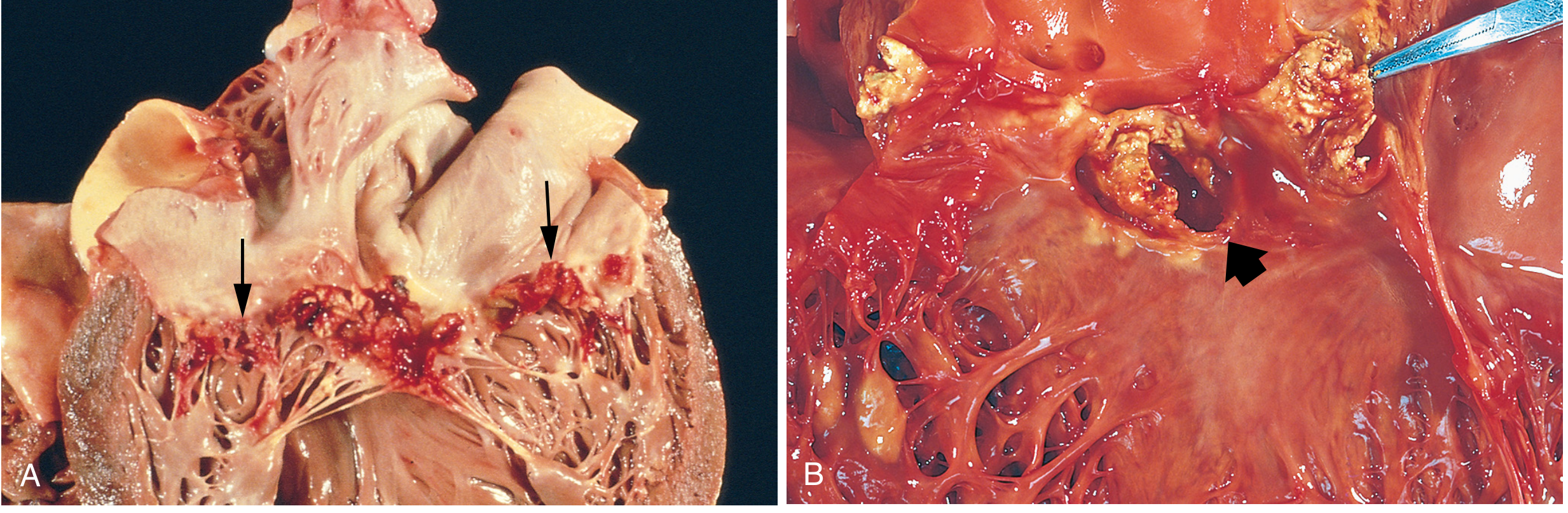
(A) Subacute mitral valve IE (Strep viridans) - large, friable vegetations on valve leaflets
(B) Acute bicuspid aortic valve IE (S. aureus) - destructive vegetations + ring abscess at annulus
Quick Comparison: Acute vs Subacute IE Morphology
| Feature | Acute IE | Subacute IE |
|---|---|---|
| Organism | S. aureus (high virulence) | Strep viridans (low virulence) |
| Valve prior state | Normal or abnormal | Pre-existing damage |
| Vegetation | Highly destructive | Less destructive |
| Base (microscopy) | No healing | Granulation tissue at base |
| Ring abscess | More common | Less common |
| Embolism | Frequent (virulent organisms) | Occurs but less violent |
Key Points from Your Notes
-
Vegetations in IE are on the atrial surface of the mitral valve and the ventricular surface of the aortic valve (the low-pressure side downstream of the regurgitant jet)
-
Embolism from left-sided IE → systemic (brain, kidneys, spleen); from right-sided → pulmonary
-
Fibrosis and calcification can develop later in both forms
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 528-529

IMG_1531.jpg
Explain
analyse_media
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
Here is a full, structured explanation of everything in your image:
Morphology of Acute Rheumatic Heart Disease (RHD)
Overview - Pancarditis
Acute rheumatic fever (RF) causes pancarditis - inflammation of ALL three layers of the heart simultaneously:
- Pericardium → Rheumatic pericarditis
- Myocardium → Rheumatic myocarditis
- Endocardium → Rheumatic endocarditis
The underlying mechanism is molecular mimicry: antibodies and CD4+ T cells against streptococcal M proteins cross-react with cardiac antigens (pericardial, myocardial, valvular). Streptococci themselves are absent from the lesions - it is entirely immune-mediated.
i. Rheumatic Pericarditis
| Feature | Detail |
|---|---|
| Gross | Fibrinous/serofibrinous exudate in pericardial sac → classic "bread and butter" appearance (fibrin strands pulled apart look like two buttered bread surfaces) |
| Microscopic | Fibrin on epicardial surface; infiltration by lymphocytes, plasma cells, histiocytes, and few neutrophils |
Bread and butter pericarditis = fibrinous strands deposited on both visceral and parietal pericardial surfaces that stick and pull apart.
ii. Rheumatic Myocarditis
Gross - Sequential Stages:
| Stage | Finding |
|---|---|
| Early | Myocardium (especially left ventricle) - soft and flabby |
| Intermediate | Interstitial tissue shows small foci of necrosis |
| Late | Tiny pale foci of Aschoff bodies scattered throughout myocardium |
Microscopic - The Aschoff Body:
The Aschoff body is the pathognomonic lesion of rheumatic fever. It is a circumscribed nodule of mixed mononuclear inflammatory cells with associated necrosis.
Components of an Aschoff body:
- Foci of lymphocytes (primarily T-cells)
- Occasional plasma cells
- Anitschkow cells (plump activated macrophages) - pathognomonic for RF
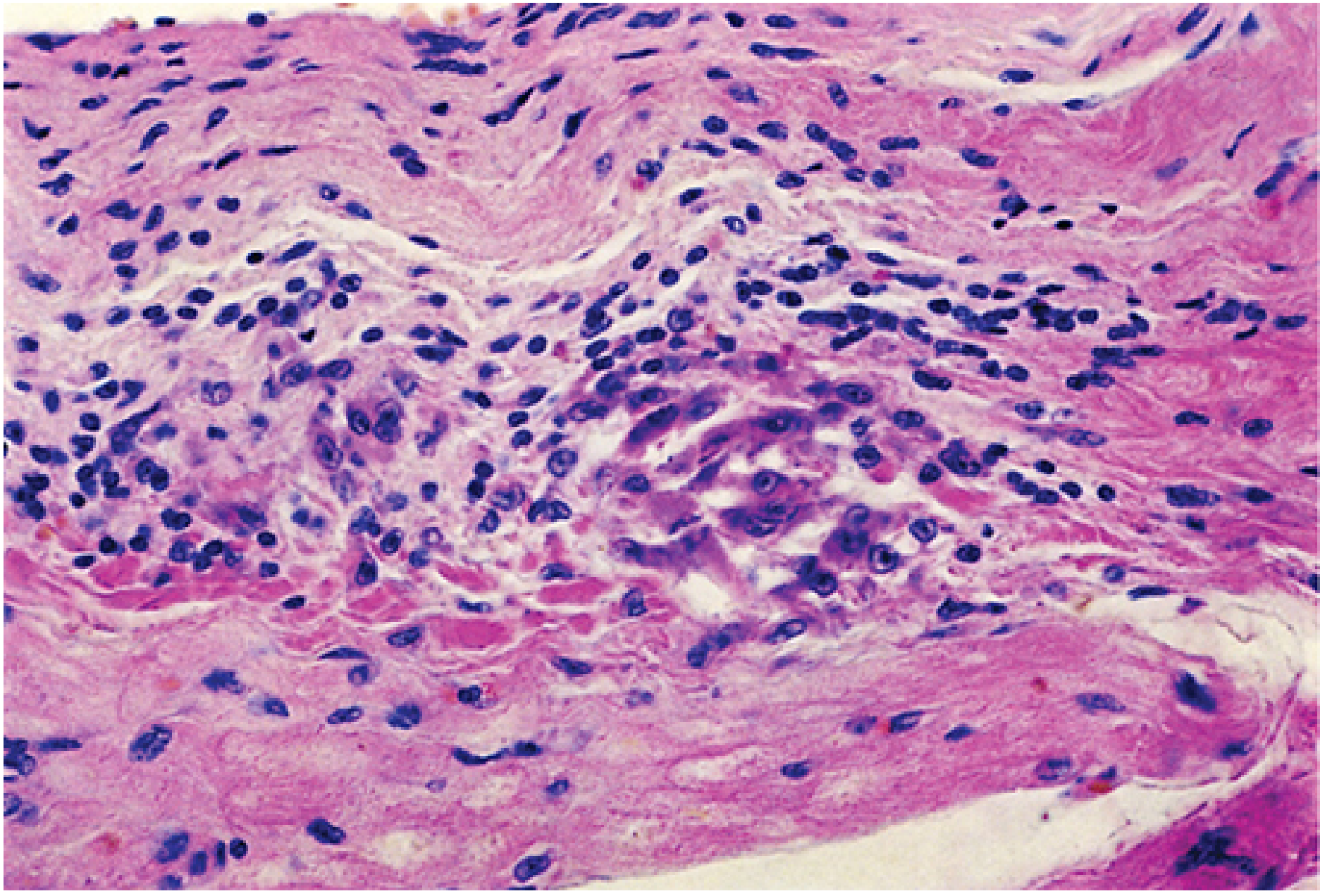
The Anitschkow Cell (must know for exams):
| Feature | Detail |
|---|---|
| Cytoplasm | Abundant |
| Nucleus | Central, round to ovoid |
| Chromatin pattern | Condensed into a central slender wavy ribbon - hence called "caterpillar cells" |
| Multinucleated form | Called Aschoff giant cells |
| Significance | Pathognomonic for Rheumatic Fever |
iii. Rheumatic Endocarditis
a. Rheumatic Valvulitis
| Feature | Detail |
|---|---|
| Gross - Vegetations | Small (1-2 mm) necrotic foci = verrucae, along the lines of valve closure |
| Valve most affected | Mitral valve (65-70%) |
| Other gross changes | Leaflet thickening, commissural fusion and shortening, thickening and fusion of tendinous cords |
| Microscopic | Edema, inflammation, and fibrinoid necrosis of valve leaflets; organization of acute inflammation → diffuse fibrosis and neovascularization → obliteration of the layered avascular leaflet architecture |
Key distinction from IE: RHD vegetations are small, sterile, firmly attached along the line of closure. IE vegetations are large, friable, and destructive.
b. Rheumatic Submural Endocarditis - MacCallum Plaques
| Feature | Detail |
|---|---|
| Cause | Regurgitant jets hit the subendocardial wall and exacerbate injury |
| Gross | Irregular thickenings on subendocardium = MacCallum plaques |
| Location | Usually the posterior wall of the left atrium |
| Microscopic | Edema, inflammatory cells, fibrinoid change of collagen |
Chronic Rheumatic Heart Disease
Valves Affected (Frequency):
- Mitral alone: 65-70%
- Mitral + Aortic: 25%
- Tricuspid + Pulmonary: Rarely
Morphology of Chronic Mitral Valve Changes:
| Change | Mechanism/Result |
|---|---|
| Leaflet thickening | Due to fibrosis from repeated attacks |
| Commissural fusion | Valve orifice becomes fixed and narrow → fish mouth / button hole appearance |
| Chord. tendinea changes | Shortening, thickening, and fusion |
| Left atrial dilation | With mural thrombi (embolism risk) |
| Right ventricular hypertrophy | Long-standing mitral stenosis → pulmonary hypertension → RVH |
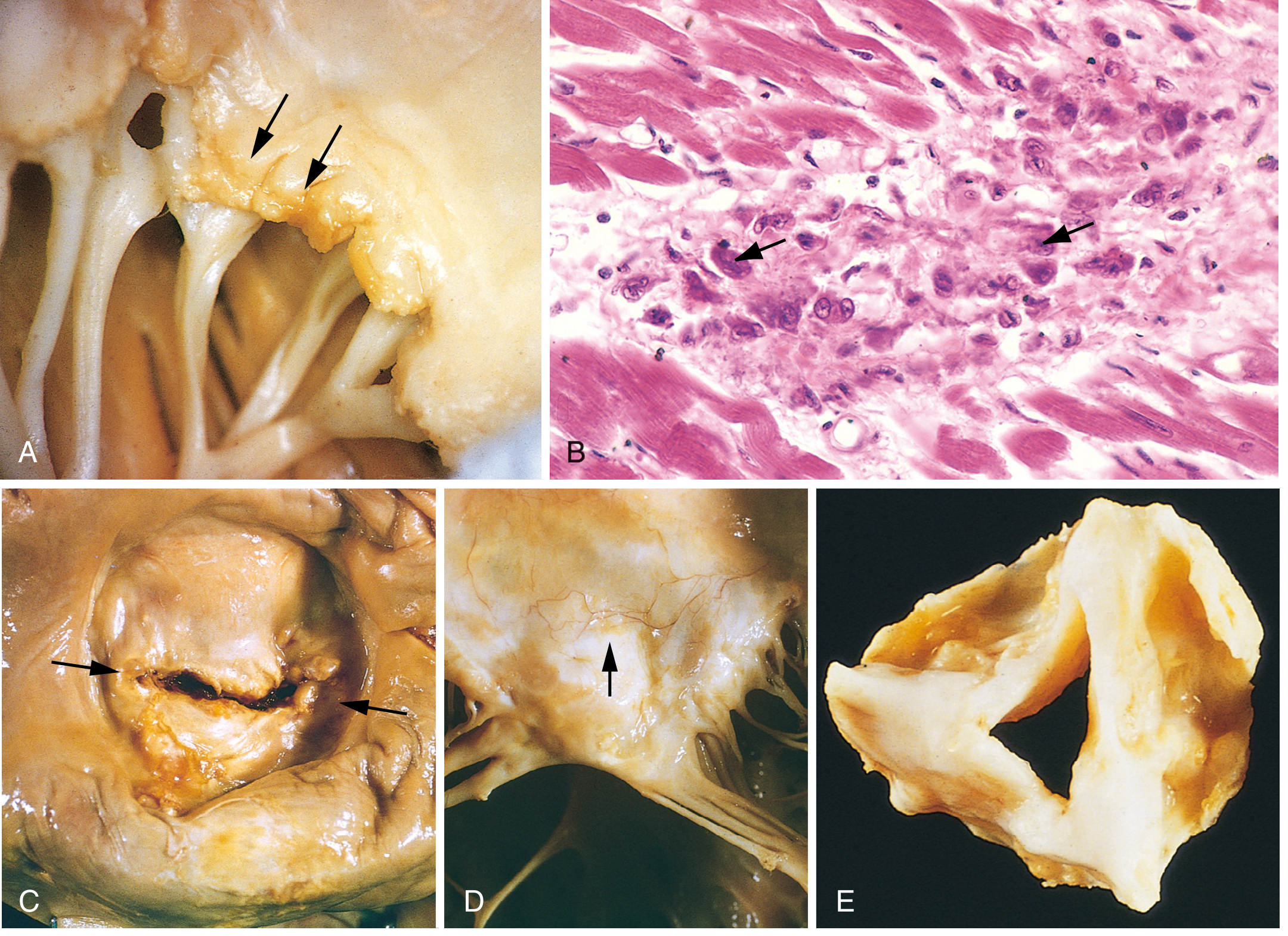
Panel A: Small warty verrucae on mitral valve leaflets (acute phase). Panel B: Aschoff body - Anitschkow cells with caterpillar chromatin (arrows). Panel C: Fish-mouth/button-hole mitral stenosis (chronic RHD). Panel D: Fused, thickened chordae. Panel E: Thickened aortic valve cusps.
Quick Memory Summary
Acute RHD = PANCARDITIS
├── Pericarditis → Bread & Butter
├── Myocarditis → Aschoff bodies (Anitschkow cells = caterpillar cells)
└── Endocarditis
├── Valvulitis → 1-2mm verrucae at lines of closure
└── Submural → MacCallum plaques (left atrium)
Chronic RHD = MITRAL STENOSIS
→ Fish mouth / Button hole orifice
→ Left atrial dilation + mural thrombi
→ Pulmonary HTN → RVH
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 525-528



IMG_1532.jpg
+2 more
Explain
analyse_media
analyse_media
Finding Sources
analyse_media
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a complete, exam-focused explanation of everything across all three images:
Myocardial Infarction (MI) - Complete Notes
Definition
MI = death of cardiac muscle due to prolonged severe ischemia.
- Ischemia lasting 20-30 minutes or longer → irreversible cell injury → MI
- The 2018 joint task force defines it as: "acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia"
Commonly Involved Vessels
| Vessel | % of MI |
|---|---|
| Left Anterior Descending (LAD) | 50% - anterior LV wall, anterior 2/3 of septum, apex |
| Right Coronary Artery (RCA) | 30-40% - right ventricle, posterior wall |
| Left Circumflex (LCA/LCX) | 15-20% - lateral LV wall |
Risk Factors
- Atherosclerosis - most common risk factor
- Age: 10% MI under 45 yrs; 45% MI under 65 yrs
- Sex: Males > Females (during reproductive years). Post-menopause, risk equalizes - protective effect of estrogen is lost
- Other: hypertension, diabetes, smoking, dyslipidemia, family history
Etiology
A. Coronary Atherosclerosis (90% of cases)
- Atherosclerotic narrowing of one or more coronary arteries
B. Non-Atherosclerotic (~10% of cases)
- Vasospasm without coronary atherosclerosis
- Emboli from:
- Left atrium (with atrial fibrillation)
- Left-sided mural thrombus
- Vegetations of infective endocarditis
- Intracardiac prosthetic material
- Paradoxical emboli - from right heart/peripheral veins through a patent foramen ovale into coronary arteries
- Other ischemic causes:
- Vasculitis
- Sickle cell disease (hematologic)
- Amyloid deposition in vascular walls
- Vascular dissection
- Lowered systemic BP (shock)
Pathogenesis
Two linked cascades:
Step 1 - Plaque Disruption → Thrombus Formation:
Atheromatous plaque: intraplaque hemorrhage / erosion / ulceration / rupture / fissuring
↓
Exposure of sub-endothelial collagen + necrotic plaque contents
↓
Platelet adherence → activation → release granule contents → aggregate → microthrombi
↓
Vasospasm (from platelet mediators: TXA2, ADP, serotonin)
↓
Activation of coagulation pathway → bulk thrombus → Arterial occlusion
Step 2 - Occlusion → Cell Death:
Severe ischemia
↓
Loss of oxidative phosphorylation → decreased ATP generation
↓
Accumulation of noxious metabolites (lactic acid, Ca²⁺ influx)
↓
Loss of contractility
↓
20-30 mins of ischemia → Irreversible cell injury
↓
Microvascular injury → Myocardial Infarction
Why does subendocardium die first? It is the most distal from epicardial vessels, receives blood last, and is exposed to highest intramural pressures. Ischemia progresses as a wavefront from subendocardium outward toward epicardium.
Types of Infarct (by Wall Thickness Involved)
| Type | Description | ECG | Cause |
|---|---|---|---|
| A. Transmural | Full-thickness; most common; along distribution of one artery | ST elevation (STEMI) | Complete epicardial occlusion |
| B. Subendocardial | Inner 1/3-1/2 of ventricular wall; least common; circumferential | Non-ST elevation (NSTEMI) | Spontaneous or therapeutic lysis before transmural necrosis |
| C. Microscopic | Small vessel occlusion (vasculitis, embolization) | No ECG changes | Small intramyocardial vessel disease |
Morphology - The Key Timeline (MOST EXAM-TESTED)
| Time | Gross Appearance | Microscopy | What's happening |
|---|---|---|---|
| 0-½ hr (reversible) | None | None | ATP loss, mitochondrial swelling only |
| ½-4 hrs | None | Wavy fibers at border | Dead fibers stretch/buckle beside contractile ones |
| 4-12 hrs | Dark mottling | Coagulation necrosis begins; edema + hemorrhage | |
| 12-24 hrs | Dark mottling | Marginal contraction band necrosis; early neutrophilic infiltrate; pyknosis of nuclei | |
| 1-3 days | Mottling with yellow-tan center | Coagulation necrosis with loss of nuclei and striations; massive neutrophilic infiltration | Peak neutrophil response |
| 3-7 days | Hyperemic border; central yellow-tan softening | Dying neutrophils; phagocytes (macrophages) appear | Macrophage phase begins |
| 7-10 days | Depressed red-tan margins | Well-developed phagocytosis; early granulation tissue at margins | Healing starts |
| 10-14 days | Red-gray depressed borders | Granulation tissue with new blood vessels + collagen deposition | Active repair |
| 2-8 weeks | Gray-white scar progressing inward | Increased collagen; decreased cellularity | Scar matures |
| >2 months | Scarring complete | Dense collagenous scar | Healed |
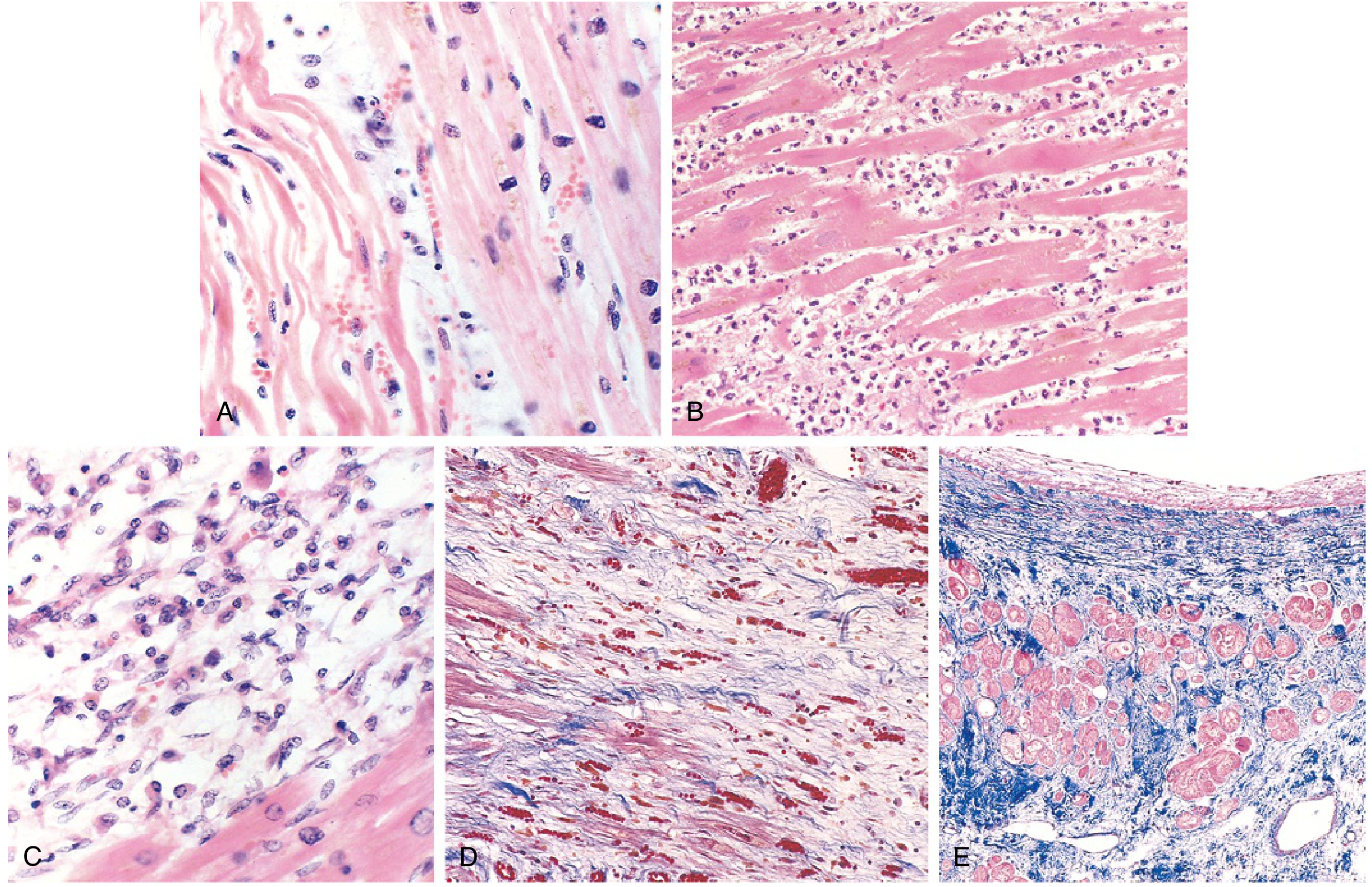
Memory aid for cellular sequence: Wavy → Necrosis → Neutrophils (peak 1-3 days) → Macrophages (3-7 days) → Granulation → Collagen → Scar
Reperfusion
Goal: Restore blood flow to ischemic myocardium to salvage muscle and limit infarct size.
Methods:
- Thrombolysis (tPA)
- Angioplasty + stent placement
- CABG (Coronary Artery Bypass Graft)
Rule: Reversible injury → Reperfusion → muscle saved. After ~20-40 min → irreversible injury.
Reperfusion Injury (the paradox - reperfusion can also cause damage):
- Mitochondrial dysfunction → membrane permeability changes → apoptosis
- Contraction band necrosis - calcium influx causes hypercontraction of sarcomeres → eosinophilic bands (hallmark of reperfused MI on microscopy)
- Free radical damage (ROS: O₂⁻, H₂O₂, •OH)
- "No-reflow" phenomenon - leukocyte aggregation in reperfused microvessels blocks blood flow
- Platelet and complement activation → further microvascular injury
Contraction band necrosis is the microscopic hallmark of reperfused MI. It is NOT seen in non-reperfused MI.
Clinical Features
| Symptom | Notes |
|---|---|
| Severe chest pain | Crushing, central, radiating to left arm/jaw |
| Dyspnea | From LV failure |
| Rapid weak pulse | Reduced CO |
| Profuse sweating (diaphoresis) | Sympathetic activation |
| Palpitations | Arrhythmias |
| Loss of consciousness | Severe pump failure or arrhythmia |
| Nausea/vomiting | Vagal activation |
| Anxiety, light-headedness, wheezing |
Investigations
| Investigation | Finding |
|---|---|
| ECG | ST elevation (STEMI) or ST depression/T-wave changes (NSTEMI) |
| Troponin T & I | Most specific cardiac marker; rises in 3-4 hrs, peaks ~24 hrs, lasts 1-2 weeks |
| CK-MB | Rises 4-8 hrs, peaks 24 hrs, returns to normal in 48-72 hrs |
| Myoglobin | Earliest marker (rises 1-2 hrs), but not cardiac-specific |
| LDH | Rises late (24-48 hrs), stays elevated for 10-14 days |
| SGOT/AST | Non-specific, rises in 8-12 hrs |
| Blood tests | Raised TLC, ESR, CRP (markers of inflammation) |
| Chest X-ray | Pulmonary congestion, cardiomegaly |
| Echocardiography | Wall motion abnormalities, EF assessment |
Complications
Severity depends on: infarct size + location + thickness (transmural vs subendocardial)
| Category | Complication | Notes |
|---|---|---|
| Contractile dysfunction | LV failure | Most common complication |
| Cardiogenic shock | In 10-15% of patients; pump failure | |
| RV failure, pulmonary congestion | ||
| Arrhythmias | Sinus bradycardia, heart block, asystole | From SA/AV node ischemia (RCA territory) |
| Tachycardia, ventricular fibrillation | Most common cause of sudden death post-MI (80-90% of early deaths) | |
| Myocardial rupture | Hemopericardium + cardiac tamponade | Occurs 3-7 days (when myocardium is soft); most lethal |
| VSD (interventricular septal rupture) | Left-to-right shunt | |
| Papillary muscle rupture | → Acute mitral regurgitation | |
| Pericarditis | Fibrinous/fibrohemorrhagic pericarditis | Develops day 2-3 after transmural MI |
| Dressler syndrome | Autoimmune pericarditis weeks later | |
| Structural | Chamber dilation, infarct expansion | Early remodeling |
| Ventricular aneurysm | Thinned wall bulges - mural thrombi form here | |
| Mural embolism | From mural thrombus in dilated/aneurysmal LV | |
| Papillary muscle dysfunction | → Mitral regurgitation (without rupture) | |
| Progressive late heart failure | Long-term LV remodeling |
Quick Summary Diagram
PLAQUE RUPTURE → THROMBUS → OCCLUSION
↓
0-½ hr: Reversible (wavy fibers)
½-4 hr: Coagulation necrosis begins
1-3 days: Neutrophil peak
3-7 days: Macrophages, softening
7-14 days: Granulation tissue
2-8 wks: Collagen scar
>2 months: Dense fibrotic scar
REPERFUSION → contraction band necrosis
COMPLICATIONS:
Early → Arrhythmias (VF), rupture (3-7d)
Mid → Pericarditis (day 2-3)
Late → Aneurysm, HF, Dressler syndrome
- Robbins & Kumar Basic Pathology, p. 353-360
analyse_media
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
Finding Sources
Finding Sources
Reading File
Here is a complete, structured explanation of everything in your image:
Vasculitis - Complete Notes
Classification of Vasculitis (by Vessel Size)
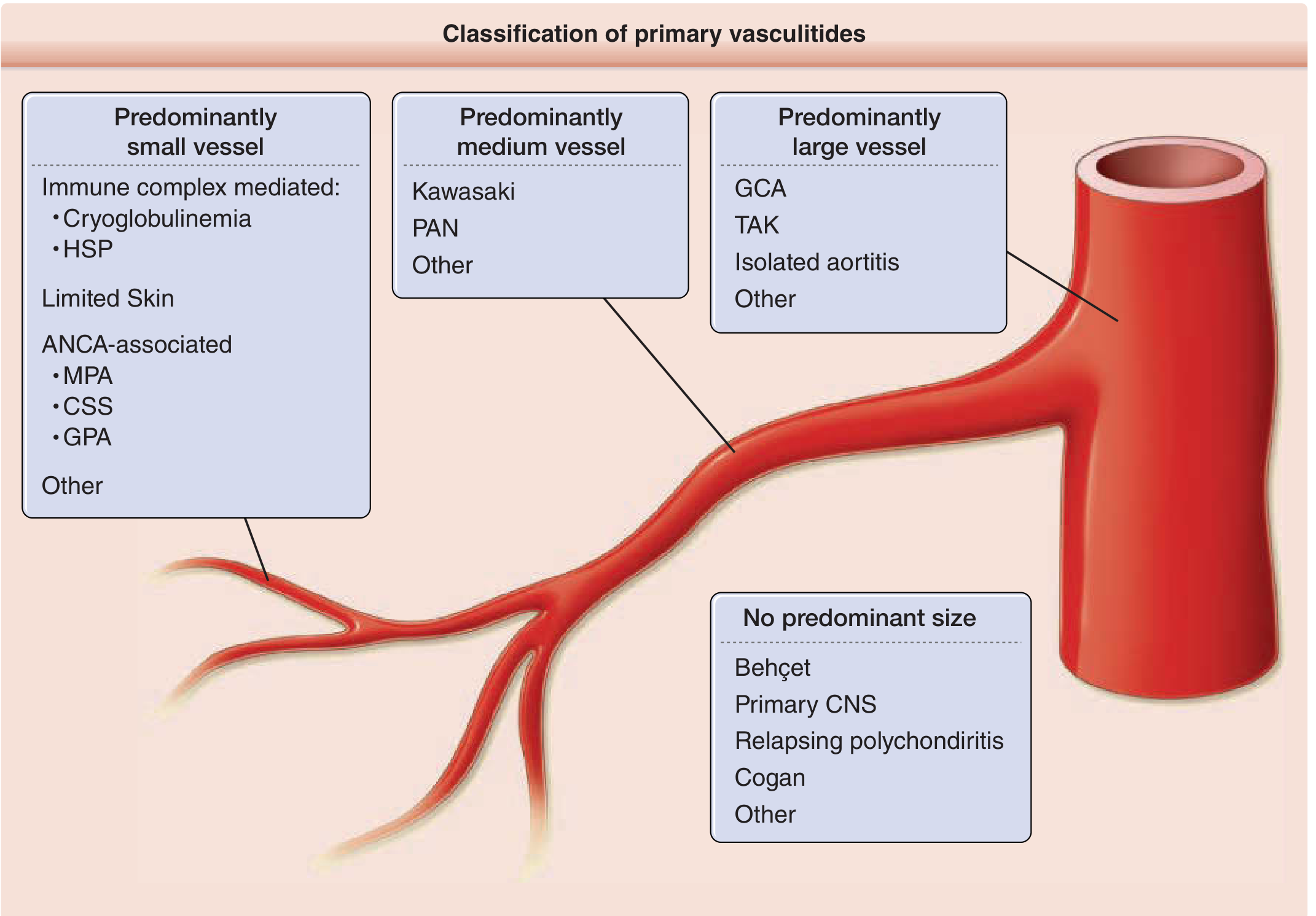
| Vessel Size | Conditions |
|---|---|
| Large vessel | Giant cell (temporal) arteritis, Takayasu arteritis |
| Medium vessel | Classic PAN, Kawasaki's disease, Buerger's disease |
| Small vessel - Immune complex mediated | SLE, Henoch-Schonlein purpura (HSP), Cryoglobulin vasculitis, Goodpasture syndrome |
| Small vessel - Paucity of immune complex (ANCA-associated) | Wegener's granulomatosis (GPA), Microscopic polyangiitis (MPA), Churg-Strauss syndrome (EGPA) |
ANCA-Associated Vasculitis
ANCA = Anti-Neutrophil Cytoplasmic Antibodies - autoantibodies against enzymes inside neutrophils. Two main types based on staining pattern:
| Feature | c-ANCA (Cytoplasmic) | p-ANCA (Perinuclear) |
|---|---|---|
| Antigen | Proteinase 3 (PR3) | Myeloperoxidase (MPO) |
| Staining pattern | Diffuse cytoplasmic | Perinuclear (around nucleus) |
| Key disease | Wegener's granulomatosis (GPA) | Microscopic polyangiitis, Churg-Strauss |
| Other p-ANCA conditions | - | Goodpasture's syndrome, Crescentic GN, Ulcerative colitis, Primary sclerosing cholangitis, Drugs (hydralazine, propylthiouracil) |
Memory: c-ANCA = PR3 = Wegener's (With cytoplasmic staining). p-ANCA = MPO = Microscopic Polyangiitis (Myeloperoxidase + Perinuclear).
Polyarteritis Nodosa (PAN)
Definition
Necrotizing vasculitis involving small and medium-sized arteries of multiple organs and tissues.
Key Features
- About 15% linked to hepatitis B viral infection; most cases are idiopathic
- Lungs are SPARED (pulmonary circulation not involved - important differentiator)
- ANCA is typically negative in classic PAN
Affected Organs (descending order)
Kidneys > Heart > Liver > GIT > Muscle > Pancreas > Testes > Nervous system > Skin
Kidney involvement → renal artery vasculitis → renin-mediated hypertension + renal infarctions Nervous system → mononeuritis multiplex in >80% of cases
Morphology
Gross:
- Involves vessel segments especially at bifurcation as tiny beaded nodules
- Aneurysmal bulges visible in gross specimens (microaneurysms)
- Lesions are segmental - skip areas present between affected segments
Microscopic - 3 Sequential Stages:
| Stage | Findings |
|---|---|
| a. Acute stage | - Fibrinoid necrosis at center of nodule in media + acute inflammatory response around necrosis - Inflammatory infiltrate in entire circumference of vessel = periarteritis - Lumen: thrombi |
| b. Healing stage | - Marked fibroblastic proliferation producing firm nodularity - Infiltrate changes to lymphocytes, plasma cells, macrophages (chronic cells) |
| c. Healed stage | - Arterial wall markedly thickened due to dense fibrosis - Internal elastic lamina fragmented or lost |
Note: Granulomatous inflammation is absent in PAN (distinguishes it from Wegener's/GPA and Churg-Strauss)
Clinical Diagnosis:
- Requires tissue biopsy OR angiogram showing microaneurysms
- Simultaneous nerve + muscle biopsy (sural nerve + gastrocnemius) has high yield
Treatment:
- Hepatitis B-associated PAN: anti-viral therapy + plasma exchange + short-course steroids
- Idiopathic PAN: high-dose prednisolone (1 mg/kg/day) → taper; cyclophosphamide for severe disease
Thromboangiitis Obliterans (Buerger's Disease)
Definition
Distinctive disease leading to vascular insufficiency - characterized by segmental, thrombosing, acute and chronic inflammation of medium-sized and small arteries, predominantly of the extremities.
Key Features:
- Affects tibial and radial arteries principally (distal vessels)
- Occasionally extends into nerves and veins of extremities
- Finally leads to gangrene of extremities
- Contains hemosiderin-laden macrophages and organized thrombus
- Most common cause of mononeuritis multiplex (per your notes)
- Develops before age of 35 in heavy smokers
- Men <40 years; higher prevalence in Asians and Eastern Europeans
Etiopathogenesis
| Cause | Mechanism |
|---|---|
| 1. Heavy cigarette smoking (main cause) | Direct endothelial toxicity OR provokes idiosyncratic immune response → hypercoagulability + thrombosis. Patients are hypersensitive to intradermally injected tobacco extracts |
| 2. Genetic thrombosis | HLA related |
Morphology
| Level | Finding |
|---|---|
| Gross | Sharply segmental acute and chronic vasculitis of medium and small arteries, predominantly extremities |
| Microscopic - Early | PMN (polymorphonuclear) infiltration of arterial walls; internal elastic lamina preserved (distinguishes from atherosclerosis) |
| Microscopic - Active | Mixed inflammatory infiltrates + luminal thrombosis; Thrombus contains micro-abscesses (neutrophils surrounded by granulomatous inflammation) |
| Microscopic - Late | Mononuclear cells + fibroblasts + giant cells replace neutrophils; inflammatory process extends to veins and nerves → encased in fibrous tissue |
| End stage | Perivascular fibrosis, organized thrombus, recanalization |
Key distinguishing feature: Internal elastic lamina is preserved in Buerger's (unlike atherosclerosis where it is destroyed). This is a major histologic differentiator.
Clinical Features
| Feature | Notes |
|---|---|
| Superficial nodular phlebitis | Migratory superficial vein thrombophlebitis |
| Cold sensitivity (Raynaud type) | In hands - vasospasm triggered by cold |
| Instep claudication | Pain in instep of foot on exercise (affects distal tibial vessels) |
| Rest pain | Severe pain even at rest - due to neural involvement |
| Chronic ulcerations | Toes, feet, fingers → Frank gangrene as disease progresses |
Clinical triad: Claudication of extremity + Raynaud phenomenon + Migratory superficial vein thrombophlebitis
Treatment:
Only effective treatment = complete tobacco abstinence. Without stopping smoking, prognosis is very poor. Arterial bypass for larger vessels, local debridement for ulcers. Amputation if above measures fail.
Quick Comparison: PAN vs Buerger's Disease
| Feature | PAN | Buerger's Disease |
|---|---|---|
| Vessel size | Small + medium | Medium + small |
| Lungs | Spared | May be involved rarely |
| Cause | Idiopathic / Hep B | Cigarette smoking |
| ANCA | Negative | Negative |
| Key microscopy | Fibrinoid necrosis, 3 stages, no granulomas | Microabscesses in thrombus, elastic lamina preserved |
| Typical patient | Any age, systemic | Young male <40, smoker |
| Treatment | Steroids ± cyclophosphamide | Stop smoking |
- Goldman-Cecil Medicine, p. (medium-vessel vasculitides chapter)
- Harrison's Principles of Internal Medicine, 22nd Edition
This is a shared conversation. Sign in to Orris to start your own chat.