Here is a comprehensive set of important pathology questions and answers for MBBS (1st/2nd year), covering all major topics from Robbins & Kumar Basic Pathology and Robbins Pathologic Basis of Disease.
Pathology - Important Questions & Answers (MBBS)
UNIT 1: CELL INJURY, DEATH & ADAPTATIONS
Q1. Define etiology and pathogenesis. How are they different?
A: Etiology is the origin/cause of a disease, including underlying causes and modifying factors (the "why"). Pathogenesis refers to the steps in the development of a disease - from the initial etiologic trigger through the cellular and molecular changes that produce the specific functional and structural abnormalities (the "how"). For example, in atherosclerosis, the etiology includes hyperlipidemia and hypertension; the pathogenesis involves endothelial injury, lipid accumulation, and foam cell formation.
- Robbins & Kumar Basic Pathology
Q2. What is homeostasis? How do cells respond to stress?
A: Homeostasis is the steady state that cells actively maintain by adjusting their structure and function to accommodate changing demands and extracellular stresses. When cells encounter physiologic stresses or injurious stimuli, they can:
- Adapt - achieve a new steady state, preserving viability (e.g., hypertrophy, hyperplasia, atrophy, metaplasia)
- Undergo reversible injury - if adaptive capability is exceeded but stimulus is mild/brief
- Die (irreversible injury) - if stress is severe or persistent
- Robbins & Kumar Basic Pathology
Q3. Enumerate the causes of cell injury.
A: Major causes:
- Hypoxia and ischemia - most common; deprive cells of oxygen (needed for ATP generation)
- Physical agents - trauma, heat/cold, radiation, electrical injury
- Chemical agents and drugs - toxins, poisons, drugs (e.g., carbon tetrachloride)
- Infectious agents - bacteria, viruses, fungi, parasites
- Immunologic reactions - autoimmune diseases, hypersensitivity reactions
- Genetic derangements - mutations causing enzyme defects, structural protein abnormalities
- Nutritional imbalances - protein-calorie malnutrition, vitamin deficiencies, excess (obesity)
- Robbins & Kumar Basic Pathology
Q4. What are the mechanisms of cell injury? Name the key biochemical targets.
A: Four key biochemical targets are affected in cell injury:
- Mitochondria - impaired oxidative phosphorylation → reduced ATP → membrane pump failure → cellular swelling
- Cellular membranes - damage to plasma and organelle membranes → loss of ion gradients, leakage of enzymes
- Nucleus (DNA) - strand breaks from free radicals or toxins → if not repaired, cell undergoes apoptosis
- Endoplasmic reticulum (ER) - accumulation of misfolded proteins triggers the unfolded protein response (UPR); severe ER stress triggers apoptosis
Irreversible injury occurs when mitochondrial damage is massive and lysosomal membranes rupture.
- Robbins & Kumar Basic Pathology
Q5. What is the difference between reversible and irreversible cell injury?
A:
| Feature | Reversible Injury | Irreversible Injury |
|---|
| Mitochondria | Swelling, loss of phospholipids | Vacuolation, large amorphous densities |
| Plasma membrane | Blebbing, blunting of microvilli | Rupture |
| Nucleus | Chromatin clumping | Pyknosis, karyorrhexis, karyolysis |
| Lysosomes | Intact | Rupture, enzymatic digestion |
| Cell fate | Returns to normal | Necrosis or apoptosis |
Key morphologic features of reversible injury: cellular swelling (most common), fatty change (steatosis), cytoplasmic vacuolization.
- Robbins & Kumar Basic Pathology
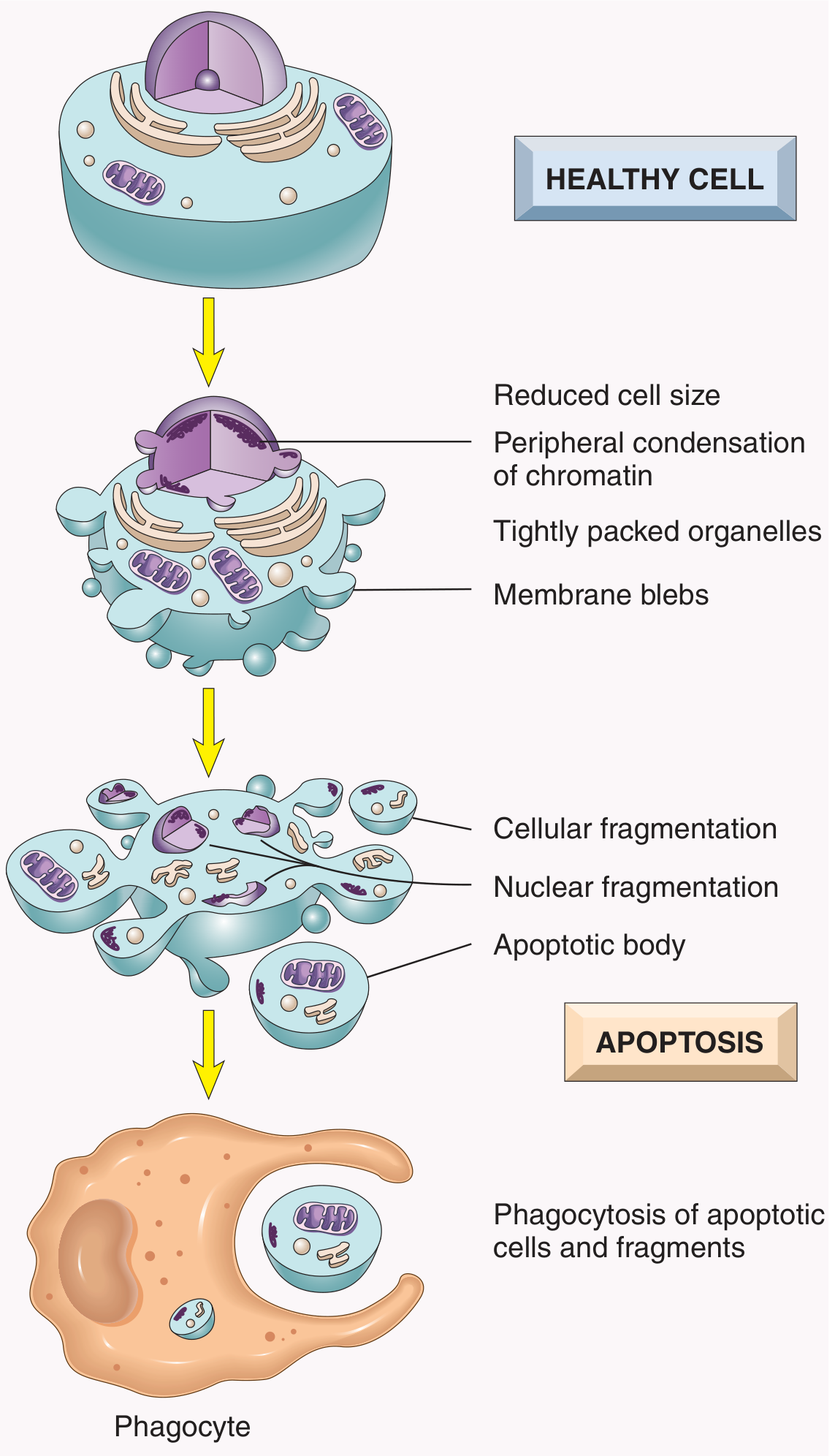
Q6. Compare necrosis and apoptosis.
A:
| Feature | Necrosis | Apoptosis |
|---|
| Cause | Ischemia, toxins, infections (pathologic) | Physiologic or pathologic |
| Cell size | Enlarged (swelling) | Reduced (shrinkage) |
| Nucleus | Pyknosis → karyorrhexis → karyolysis | Fragmentation into nucleosome-sized pieces |
| Plasma membrane | Disrupted | Intact, blebbing |
| Cell contents | Leaked - causes inflammation | Packaged in apoptotic bodies, no inflammation |
| Adjacent inflammation | Yes (prominent) | No |
| Examples | MI, ischemic stroke | Embryogenesis, immune cell deletion, hormone-deprived tissue |
Importantly, these two pathways may overlap - apoptosis can progress to necrosis ("secondary necrosis").
- Robbins & Kumar Basic Pathology, Robbins Pathologic Basis of Disease
Q7. What are the morphologic patterns of necrosis? Give examples.
A:
| Pattern | Mechanism | Example |
|---|
| Coagulative | Protein denaturation preserves cell outlines | Myocardial infarction, kidney infarction |
| Liquefactive | Enzymatic digestion liquefies tissue | Brain infarction, abscess |
| Caseous | Combination: cheese-like, structureless | Tuberculosis (TB) |
| Fat necrosis | Lipase digestion of fat | Acute pancreatitis, breast trauma |
| Fibrinoid | Immune complexes + fibrin deposition in vessel walls | Vasculitis, malignant hypertension |
| Gangrenous | Usually coagulative; "wet" gangrene has superimposed liquefaction | Diabetic foot, bowel infarction |
- Robbins & Kumar Basic Pathology
Q8. What are the cellular adaptations to stress? Define each.
A:
- Hypertrophy - increase in cell size (not number). Occurs in non-dividing cells (cardiac muscle, skeletal muscle). E.g., cardiac hypertrophy in hypertension.
- Hyperplasia - increase in cell number. Occurs in dividing cells. E.g., endometrial hyperplasia with estrogen excess.
- Atrophy - decrease in cell size/number. E.g., muscle wasting from disuse, denervation.
- Metaplasia - replacement of one differentiated cell type by another, usually more resistant type. E.g., squamous metaplasia of bronchial epithelium in smokers (columnar → squamous). Reversible but predisposes to malignancy.
- Dysplasia - disordered growth; considered pre-malignant. E.g., cervical dysplasia (CIN).
- Robbins & Kumar Basic Pathology
Q9. What is intracellular accumulation? Give examples.
A: When cells are incapable of metabolizing or exporting a substance, it accumulates. Types:
- Steatosis (fatty change) - triglycerides in liver cells; caused by alcohol, obesity, diabetes, toxins
- Cholesterol accumulation - atherosclerosis, xanthomas (foam cells)
- Protein accumulation - Russell bodies in plasma cells (immunoglobulins)
- Hyaline change - intracellular pink, glassy material (e.g., alcoholic hyaline/Mallory bodies in hepatocytes)
- Glycogen accumulation - glycogen storage diseases (e.g., von Gierke disease - glucose-6-phosphatase deficiency)
- Pigments - lipofuscin ("wear and tear" pigment, brown), melanin, hemosiderin (iron), carbon (anthracosis)
- Pathologic calcification - dystrophic (in dead tissue, normal serum calcium) vs. metastatic (in normal tissue, elevated serum calcium)
- Robbins & Kumar Basic Pathology
UNIT 2: INFLAMMATION & REPAIR
Q10. Define inflammation. What are its cardinal signs?
A: Inflammation is a response of vascularized tissues to infections, tissue damage, and other injurious stimuli. Its purpose is to eliminate the cause of cell injury, clear necrotic debris, and initiate tissue repair.
Cardinal signs (Celsus/Virchow):
- Rubor (redness) - increased blood flow
- Calor (heat) - increased blood flow
- Tumor (swelling) - increased vascular permeability
- Dolor (pain) - prostaglandins and bradykinin
- Functio laesa (loss of function) - Virchow's addition
- Robbins & Kumar Basic Pathology
Q11. Compare acute and chronic inflammation.
A:
| Feature | Acute | Chronic |
|---|
| Onset | Fast (minutes to days) | Slow (weeks to months) |
| Duration | Short | Prolonged |
| Key cells | Neutrophils | Lymphocytes, macrophages, plasma cells |
| Vascular changes | Prominent | Less prominent |
| Tissue injury | Mild, often reversible | Prominent, fibrosis |
| Causes | Infections, trauma | Persistent infections (TB, fungal), autoimmune, foreign material |
| Outcome | Resolution, abscess, or progression | Scarring/fibrosis |
Q12. What are the mediators of inflammation? Classify them.
A:
Cell-derived mediators:
- Histamine - first mediator released; from mast cells and basophils; causes vasodilation and increased permeability
- Prostaglandins - from arachidonic acid via COX pathway; cause vasodilation, pain, fever
- Leukotrienes - from arachidonic acid via lipoxygenase; LTB4 is chemotactic; LTC4/LTD4 increase permeability and cause bronchoconstriction
- Cytokines - TNF, IL-1 (from macrophages): systemic effects (fever, acute phase proteins); IL-8: chemotaxis
- PAF (Platelet Activating Factor) - bronchoconstriction, chemotaxis
- Nitric Oxide (NO) - vasodilation, kills microbes
Plasma-derived mediators (produced in liver, circulate as inactive precursors):
-
Complement (C3a, C5a) - C5a is most important: chemotaxis, opsonization, MAC formation
-
Kinin system - bradykinin: pain, vasodilation, increased permeability
-
Coagulation system - fibrin, thrombin
-
Robbins & Kumar Basic Pathology
Q13. What is a granuloma? What are its causes?
A: A granuloma is a focal accumulation of activated macrophages (epithelioid cells), often surrounded by lymphocytes, with or without giant cells and central necrosis. It is the hallmark of chronic granulomatous inflammation.
Components:
- Epithelioid cells (activated macrophages)
- Langhans giant cells (horseshoe-shaped nuclei) or foreign-body giant cells
- Peripheral lymphocytes
- Central caseous necrosis (in TB)
Causes (mnemonic: "SATCH-B"):
- S - Sarcoidosis (non-caseating)
- A - Allergic alveolitis
- T - Tuberculosis (caseating - most classic)
- C - Crohn's disease
- H - Histoplasmosis / other fungi
- B - Berylliosis, foreign body reaction, leprosy (syphilis)
Q14. What is wound healing? Describe primary vs. secondary intention.
A:
Wound healing involves:
- Hemostasis - platelet aggregation, fibrin clot
- Inflammation - neutrophils then macrophages clean debris
- Proliferation - angiogenesis, fibroblast proliferation, collagen synthesis
- Remodeling - collagen cross-linking, scar maturation
Primary intention (1° intention): Clean incised wound with opposed edges. Minimal tissue loss. Narrow scar. E.g., surgical incision.
Secondary intention (2° intention): Wound with large tissue defect. Requires more granulation tissue, greater contraction, larger scar. E.g., abscess cavity, extensive burns.
Factors impairing healing: Infection, malnutrition (vitamin C deficiency - impairs collagen synthesis), diabetes (poor vascularization, immunity), steroids (inhibit inflammation and collagen synthesis), poor blood supply.
UNIT 3: HEMODYNAMICS, THROMBOSIS & SHOCK
Q15. What is thrombosis? What is Virchow's triad?
A: Thrombosis is the formation of a solid mass (thrombus) from blood components within the vasculature in a living individual.
Virchow's Triad (the three predisposing factors):
- Endothelial injury - most important in arterial thrombosis; exposes subendothelial collagen → platelet activation. Causes: atherosclerosis, hypertension, vasculitis.
- Abnormal blood flow (turbulence/stasis) - prevents dilution of activated clotting factors, reduces laminar flow (keeps platelets away from endothelium). Turbulence causes arterial/cardiac thrombi; stasis causes venous thrombi.
- Hypercoagulability - primary (genetic: Factor V Leiden, prothrombin mutation, antithrombin III deficiency) or secondary (acquired: prolonged bed rest, cancer, pregnancy, OCP use).
Q16. What is embolism? What are the types?
A: An embolism is the passage of a detached intravascular mass (embolus) from its site of origin to a distant site.
Types:
- Thromboembolism - most common (95%+); DVT → pulmonary embolism (PE) most common
- Fat embolism - fractures of long bones, liposuction; fat globules obstruct pulmonary/cerebral vessels
- Air embolism - IV injections, surgery; >100 mL can be fatal
- Amniotic fluid embolism - obstetric complication; can cause DIC and respiratory failure
- Tumor embolism - cancer cells spread via hematogenous route
- Paradoxical embolism - embolus crosses from venous to arterial system through a patent foramen ovale
Q17. Define infarction. What factors determine outcome?
A: Infarction is an area of ischemic coagulative necrosis caused by occlusion of the vascular supply (arterial or venous).
Types:
- White (anemic) infarct - solid organs with single blood supply (heart, kidney, spleen). Wedge-shaped.
- Red (hemorrhagic) infarct - loose tissue (lung, intestine) or organs with dual blood supply; also in venous occlusions.
Factors determining outcome:
- Nature of vascular supply (single vs. dual; end artery vs. collateral)
- Rate of occlusion (slow → collaterals can develop)
- Tissue vulnerability to hypoxia (neurons: 3-5 min; myocardium: 20-30 min; fibroblasts: hours)
- Oxygen-carrying capacity of blood (anemia worsens outcome)
Q18. What are the types of shock? What is the mechanism of each?
A:
| Type | Mechanism | Examples |
|---|
| Cardiogenic | Pump failure → reduced cardiac output | MI, cardiac tamponade, arrhythmia |
| Hypovolemic | Loss of blood/fluid volume | Hemorrhage, burns, severe diarrhea |
| Distributive (Septic) | Systemic vasodilation → maldistribution of flow | Gram-negative sepsis (endotoxin → NO release) |
| Neurogenic | Loss of vascular tone (autonomic dysfunction) | Spinal cord injury, general anesthesia |
| Anaphylactic | IgE-mediated mast cell degranulation → massive vasodilation | Bee sting, drug allergy |
Stages of shock: (1) Compensated (nonprogressive) → (2) Progressive → (3) Irreversible
UNIT 4: NEOPLASIA
Q19. Define neoplasia. How is a benign tumor different from a malignant tumor?
A: A neoplasm is an abnormal mass of tissue showing uncoordinated growth that exceeds that of normal tissues and persists after the stimulus causing it ceases (Willis's definition). Neoplasia literally means "new growth."
| Feature | Benign | Malignant |
|---|
| Differentiation | Well differentiated | Poorly differentiated to anaplastic |
| Growth rate | Slow | Rapid |
| Invasion | No invasion | Locally invasive |
| Metastasis | Never | Yes (hallmark of malignancy) |
| Border | Encapsulated, well-defined | Irregular, infiltrating |
| Necrosis/hemorrhage | Rare | Common |
| Mitoses | Rare, normal | Frequent, atypical |
| Nuclear features | Normal N:C ratio | High N:C ratio, hyperchromatism, prominent nucleoli |
Q20. What are proto-oncogenes, oncogenes, and tumor suppressor genes?
A:
Proto-oncogenes are normal cellular genes that regulate cell growth, differentiation, and death. They become oncogenes when mutated or overexpressed, driving uncontrolled cell proliferation.
Examples:
- RAS - most commonly mutated oncogene in human cancers; point mutation → constitutively active GTPase
- MYC - transcription factor; amplified in Burkitt lymphoma (t8;14 translocation)
- HER2/neu (ERBB2) - amplified in ~25% breast cancers; target of trastuzumab
- BCR-ABL - Philadelphia chromosome (t9;22); tyrosine kinase; CML; target of imatinib
Tumor suppressor genes (anti-oncogenes) normally brake cell proliferation. Loss of function (both alleles) allows uncontrolled growth ("two-hit hypothesis" - Knudson):
- RB (retinoblastoma gene) - first tumor suppressor described; mutated in retinoblastoma, osteosarcoma
- TP53 - "guardian of the genome"; most commonly mutated tumor suppressor; mutated in >50% of all human cancers; Li-Fraumeni syndrome (germline p53 mutation)
- APC - mutated in familial adenomatous polyposis (FAP); loss → accumulation of β-catenin
- BRCA1/BRCA2 - DNA repair genes; mutations → breast and ovarian cancer
Q21. What are the routes of metastasis? Which tumors spread by each route?
A:
-
Lymphatic spread - most common for carcinomas (epithelial tumors). Sentinel lymph node is the first draining node. E.g., breast cancer → axillary nodes; colon cancer → mesenteric nodes.
-
Hematogenous spread - characteristic of sarcomas (connective tissue tumors) but also carcinomas. Travels via veins (portal → liver; systemic veins → lungs; vertebral venous plexus → spine - "Batson plexus").
- Liver - most common site of metastasis overall; GI cancers via portal vein
- Lungs - most common for cancers draining into systemic veins
- Bone - breast, prostate, lung, thyroid, kidney ("BPro-LTK")
- Brain - lung, breast, melanoma, kidney, colon
-
Seeding of body cavities (transcoelomic) - implantation on peritoneal/pleural surfaces. E.g., ovarian cancer → peritoneal seeding ("Krukenberg tumor" = ovarian metastasis from GI cancer via transcoelomic or hematogenous spread).
Q22. What is the role of p53 in cancer?
A: p53 (encoded by TP53) is a transcription factor and the most important tumor suppressor in humans, mutated in >50% of all cancers.
Functions of normal p53:
- DNA damage sensor - activated by DNA strand breaks, hypoxia, oncogene activation
- Cell cycle arrest - induces p21 (CDK inhibitor) → G1/S checkpoint arrest → allows DNA repair
- Apoptosis - if DNA damage is unrepairable, p53 activates BAX → apoptosis
- Senescence - permanent cell cycle arrest
- Inhibits angiogenesis - promotes TSP-1 expression
Consequences of p53 loss:
- Cells with damaged DNA continue to replicate → accumulation of mutations → cancer
- Li-Fraumeni syndrome: germline TP53 mutation → multiple cancers at young age
Q23. What is grading and staging of tumors? Why is staging more important clinically?
A:
Grading refers to the histologic assessment of tumor differentiation (how much it resembles normal tissue):
- Grade I: Well differentiated
- Grade II: Moderately differentiated
- Grade III: Poorly differentiated
- Grade IV: Anaplastic/undifferentiated
Staging refers to the extent of tumor spread, assessed clinically and pathologically using the TNM system:
- T = Tumor size/local extent (T1-T4)
- N = Regional lymph node involvement (N0-N3)
- M = Distant metastasis (M0 or M1)
Staging is more important clinically because it directly determines prognosis and treatment (e.g., surgery vs. chemotherapy vs. palliative care). Grading helps select therapy but varies in reliability.
UNIT 5: IMMUNITY & HYPERSENSITIVITY
Q24. Classify hypersensitivity reactions (Gell and Coombs classification).
A:
| Type | Name | Mechanism | Mediators | Examples |
|---|
| Type I | Immediate / Anaphylactic | IgE on mast cells → allergen crosslinks IgE → degranulation | Histamine, leukotrienes, prostaglandins | Anaphylaxis, asthma, urticaria, hay fever |
| Type II | Cytotoxic / Antibody-dependent | IgG/IgM bind cell surface antigens → complement activation or ADCC | Complement, NK cells | Hemolytic disease of newborn, Goodpasture's, autoimmune hemolytic anemia, myasthenia gravis |
| Type III | Immune complex | Antigen-antibody complexes deposited in tissues → complement → neutrophil recruitment | Complement, neutrophils | SLE, serum sickness, post-streptococcal GN, polyarteritis nodosa |
| Type IV | Delayed-type (DTH) / Cell-mediated | CD4+ T cells (Th1) or CD8+ CTLs react to antigen 24-72 hrs later | IFN-γ, TNF, cytotoxic T cells | Contact dermatitis, TB skin test (Mantoux), graft rejection, type 1 DM |
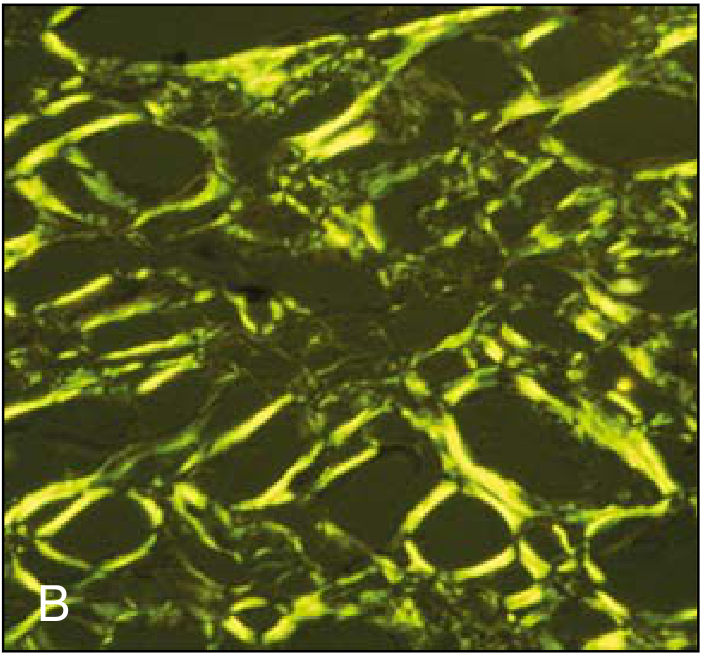
Q25. What is amyloidosis? How is it classified?
A: Amyloidosis is a pathologic extracellular deposition of insoluble abnormal fibrillar proteins that form beta-pleated sheets. It stains Congo red and shows apple-green birefringence under polarized light.
Classification:
- Primary (AL) amyloidosis - light chains from plasma cell dyscrasia (multiple myeloma). Most common type. Affects heart, kidney, tongue, nerves.
- Secondary (AA) amyloidosis - serum amyloid A (SAA), an acute phase reactant, in chronic inflammatory diseases (TB, rheumatoid arthritis, IBD, osteomyelitis). Affects kidney (most common organ), liver, spleen.
- Hereditary (familial) amyloidosis - mutant transthyretin (TTR). E.g., familial Mediterranean fever → AA type.
- Dialysis-related - β2-microglobulin accumulates in long-term dialysis patients (carpal tunnel syndrome).
- Localized - e.g., Alzheimer disease (Aβ amyloid in brain plaques), type 2 DM (islet amyloid polypeptide in pancreatic islets).
Key organ effects: Kidney → nephrotic syndrome; Heart → restrictive cardiomyopathy; Spleen → "sago" or "lardaceous" spleen.
UNIT 6: SELECTED ORGAN-SPECIFIC PATHOLOGY
Q26. What are the morphologic features of myocardial infarction (MI) at different time intervals?
A:
| Time | Gross | Microscopic |
|---|
| 0-4 hrs | Normal | Normal (no changes) |
| 4-12 hrs | Pallor begins | Wavy fibers, coagulation necrosis begins |
| 12-24 hrs | Pallor | Coagulative necrosis, loss of nuclei/striations, neutrophil infiltration begins |
| 1-3 days | Yellow pallor with hyperemic border | Dense neutrophilic infiltrate |
| 3-7 days | Hyperemic border, central yellow-white | Macrophage infiltration, removal of dead cells |
| 1-3 weeks | Fibrovascular granulation tissue | Granulation tissue replaces necrotic muscle |
| >6 weeks | White fibrous scar | Dense collagen scar |
Most common site: Left anterior descending artery (LAD) → anterior wall and septum of LV.
Q27. What are the types of hepatitis and their key features?
A:
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|
| Transmission | Fecal-oral | Parenteral, sexual, perinatal | Parenteral (IV drug use #1) | Parenteral (needs HBV) | Fecal-oral |
| Chronicity | None | 5-10% | 80% | Yes (with HBV) | None |
| Cirrhosis | No | Yes | Yes (most common cause in West) | Yes | No |
| HCC association | No | Yes (HBsAg carrier) | Yes | Yes | No |
| Vaccine | Yes | Yes | No | HBV vaccine prevents HDV | None widely available |
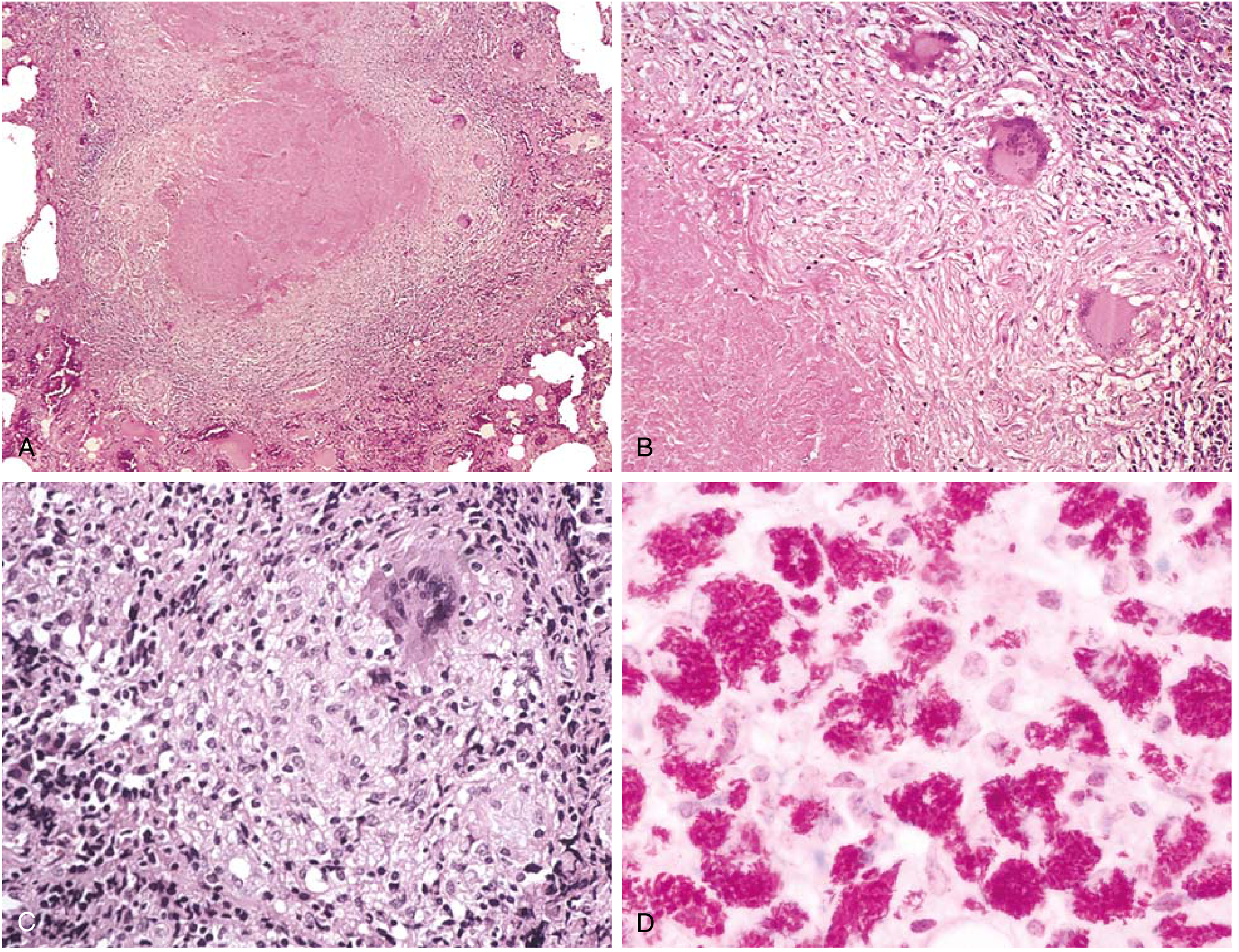
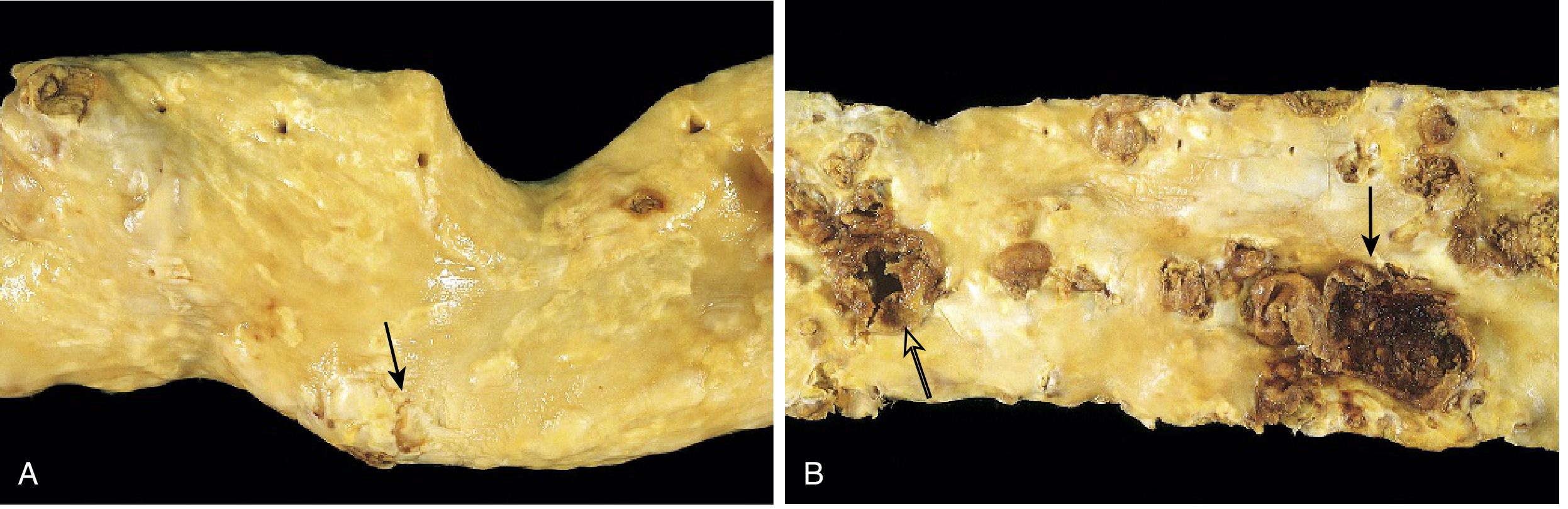
Q28. What is the pathology of tuberculosis? Describe primary vs. secondary TB.
A:
Primary TB:
- First exposure, usually in children
- Subpleural focus (Ghon focus) + hilar lymph node involvement = Ghon complex
- Usually asymptomatic; heals with calcification
- Rarely: progressive primary TB or miliary TB in immunocompromised
Secondary (reactivation) TB:
- Reactivation of latent primary infection (usually >90% of adult cases)
- Usually upper lobe of lung (high O2 tension)
- Hallmark: caseating granuloma with central cheese-like necrosis
- Complications: cavity formation, bronchiectasis, massive hemoptysis, miliary TB, CNS TB, Pott's disease (vertebral TB), TB lymphadenitis
Key histology: Epithelioid cell granuloma + Langhans giant cells + caseous necrosis + peripheral lymphocytes
Test: Mantoux/PPD (tuberculin skin test) = type IV hypersensitivity; positive >10 mm induration at 48-72 hrs
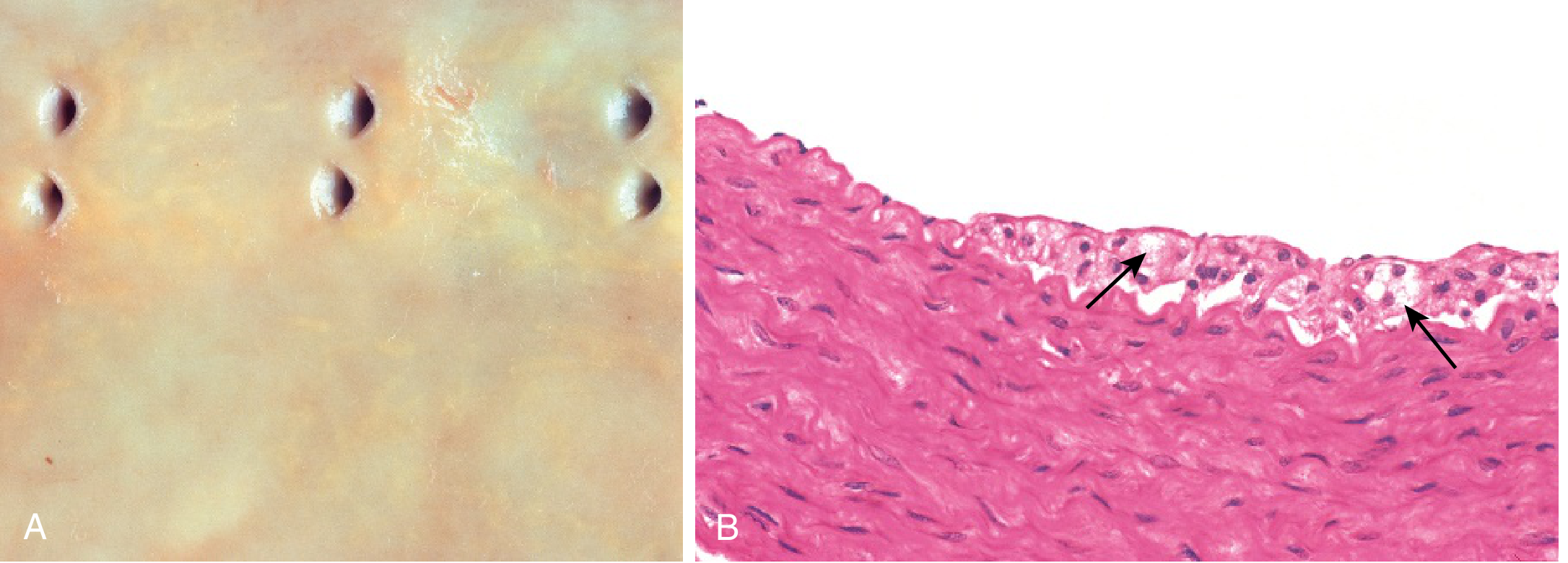
Q29. What is the pathogenesis of atherosclerosis?
A: The Response to Endothelial Injury hypothesis (Ross):
- Endothelial injury (from hypertension, hyperlipidemia, smoking, homocysteine) → endothelial dysfunction
- Increased permeability → LDL enters intima → oxidized LDL (ox-LDL)
- Monocyte recruitment → monocytes adhere to endothelium (via VCAM-1, ICAM-1) → enter intima → macrophages → engulf ox-LDL → foam cells
- Fatty streak - earliest visible lesion (reversible); foam cells + T lymphocytes
- Platelet aggregation → smooth muscle migration from media to intima → proliferation + ECM production
- Fibrous plaque - necrotic core (foam cells, cellular debris, cholesterol crystals) + fibrous cap (SMC + collagen) → advanced atheroma
- Plaque rupture/erosion → thrombosis → acute MI or stroke
Risk factors (major): hyperlipidemia (LDL), hypertension, smoking, diabetes, family history.
Q30. What is carcinoma of the cervix? How does HPV cause it?
A:
Cervical carcinoma is the most common gynecologic malignancy worldwide. Most commonly squamous cell carcinoma (80%); adenocarcinoma (15-20%).
HPV role:
- High-risk HPV types (16, 18, 31, 33) - responsible for >95% of cervical cancers
- HPV E6 protein → binds and degrades p53
- HPV E7 protein → binds and inactivates pRb (retinoblastoma protein)
- This releases E2F transcription factor → uncontrolled cell cycle progression
Progression: Normal → CIN I (mild dysplasia) → CIN II (moderate) → CIN III/CIS (severe/carcinoma in situ) → invasive carcinoma
Koilocytes (HPV-infected cells with perinuclear halo and wrinkled nucleus) are the hallmark on Pap smear.
Screening: Pap smear (cervical cytology) + HPV DNA testing
Prevention: HPV vaccines (Gardasil/Cervarix) - protect against HPV 16, 18 (and types 6, 11 for genital warts)
UNIT 7: HEMATOLOGY BASICS
Q31. How are anemias classified? Give examples of each.
A:
Morphological classification (by RBC size/color):
- Microcytic hypochromic - iron deficiency anemia (most common), thalassemia, sideroblastic anemia, anemia of chronic disease
- Macrocytic (normochromic) - megaloblastic (B12/folate deficiency), liver disease, hypothyroidism, drugs (hydroxyurea)
- Normocytic normochromic - hemolytic anemia, aplastic anemia, anemia of chronic disease (early), blood loss (acute)
Pathophysiologic classification:
- Blood loss (acute vs. chronic)
- Increased destruction (hemolytic) - intrinsic (sickle cell, G6PD deficiency, hereditary spherocytosis) or extrinsic (autoimmune, microangiopathic)
- Decreased production - iron deficiency, B12/folate deficiency, aplastic anemia, renal failure
Q32. What is sickle cell disease? What is its pathogenesis and complications?
A:
Mutation: Single nucleotide substitution in the beta-globin gene (GAG → GTG), causing glutamate → valine at position 6. This creates HbS.
Pathogenesis: On deoxygenation, HbS polymerizes → RBCs sickle → increased viscosity + occlusion of small vessels + hemolysis.
Triggers of sickling: Hypoxia, acidosis, dehydration, infection, cold.
Complications:
- Vaso-occlusive crises - pain crises (bones, chest, abdomen)
- Acute chest syndrome - fever + pulmonary infiltrates; most common cause of death in adults
- Stroke - especially in children
- Splenic sequestration/autosplenectomy - functional asplenia → susceptibility to encapsulated organisms (Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis)
- Aplastic crisis - triggered by Parvovirus B19 infection
- Avascular necrosis - especially femoral head
- Renal papillary necrosis
Diagnosis: Hemoglobin electrophoresis (gold standard)
Protection: HbF (fetal Hb) inhibits sickling. Hydroxyurea increases HbF levels.
QUICK RECALL TABLE: KEY STAINS IN PATHOLOGY
| Stain | What it identifies |
|---|
| H&E | Routine histology |
| PAS (Periodic Acid-Schiff) | Glycogen, fungi (Cryptococcus, Histoplasma), basement membranes |
| Congo red | Amyloid (apple-green birefringence under polarized light) |
| Ziehl-Neelsen (ZN/AFB) | Acid-fast bacilli (M. tuberculosis, M. leprae) |
| Gram stain | Bacteria (gram +ve = purple; gram -ve = pink) |
| Sudan IV / Oil Red O | Lipids/fat (frozen sections) |
| Prussian Blue (Perl's) | Hemosiderin (iron) |
| Silver stain (Gomori) | Fungi, Pneumocystis, reticulin fibers |
| India ink | Cryptococcus capsule |
| Giemsa | Malaria, Leishmania, chromosomes |
| Masson's Trichrome | Collagen (blue), muscle (red) |
QUICK RECALL: IMPORTANT TUMOR MARKERS
| Tumor Marker | Cancer |
|---|
| PSA | Prostate |
| AFP (alpha-fetoprotein) | Hepatocellular carcinoma, germ cell tumors (yolk sac) |
| β-hCG | Choriocarcinoma, hydatidiform mole |
| CEA | Colorectal cancer (also pancreas, lung, breast) |
| CA-125 | Ovarian cancer |
| CA 19-9 | Pancreatic cancer |
| CA 15-3 | Breast cancer |
| S-100 | Melanoma, neural tumors |
| Calcitonin | Medullary thyroid carcinoma |
| Chromogranin A | Neuroendocrine tumors |
Sources: Robbins & Kumar Basic Pathology (10th ed.) and Robbins, Cotran & Kumar Pathologic Basis of Disease - accessed via the medical textbook library.
This covers all the major topics examined in MBBS pathology. If you want deeper coverage on any specific topic (e.g., renal pathology, CNS tumors, lymphomas, lung pathology, GI diseases), just ask and I'll expand those sections.