epilepsy
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"epilepsy"[MeSH Terms] AND "treatment"[MeSH Terms]
Finding Sources
Searching PubMed
"epilepsy" AND "antiepileptic drugs"
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the full response.
Epilepsy: Management & Treatment
Sources: Harrison's Principles of Internal Medicine 22E (2025); Bradley & Daroff's Neurology in Clinical Practice; Swanson's Family Medicine Review
1. General Principles
The goal of treatment is complete seizure control with minimal adverse effects. Because individual responses to antiseizure medications (ASMs) are unpredictable, therapy involves trial and error — a process that may take months. Monotherapy should always be the goal. Most ASMs require gradual titration to minimize side effects.
Key monitoring principles:
- Serum drug levels guide initial dosing but are not the primary target — clinical response and tolerability are
- Free (unbound) drug levels are more relevant than total levels, especially in patients with hypoalbuminemia (hepatic or renal disease)
- Monitor for compliance, toxicity, and baseline levels before pregnancy
2. Antiseizure Drug Selection by Seizure Type
Focal (Partial) Seizures
| Drug | Notes |
|---|---|
| Lamotrigine | Best time-to-treatment-failure in community trials; requires slow titration |
| Carbamazepine / Oxcarbazepine | Carbamazepine is a classic first-line agent; OXC can be started at effective dose |
| Levetiracetam | Can be started at therapeutic dose immediately; good when rapid onset needed |
| Eslicarbazepine, lacosamide, brivaracetam, zonisamide, tiagabine, perampanel | Second-line or adjunctive for focal seizures |
| Cenobamate | Recently approved; significantly improves seizure control after failure of ≥3 drugs |
| Topiramate | Effective but requires slow titration; cognitive adverse effects limit first-line use |
Lamotrigine was significantly superior to carbamazepine, gabapentin, and topiramate for time-to-treatment failure in the SANAD trial (Marson et al., 2007). When rapid effect is needed, oxcarbazepine or levetiracetam are drugs of choice.
Generalized Seizures
| Seizure Type | First-Line Drug(s) | Notes |
|---|---|---|
| Primary generalized tonic-clonic (GTC) | Lamotrigine, valproic acid, levetiracetam | Carbamazepine, oxcarbazepine, phenytoin can worsen generalized seizures |
| Absence | Ethosuximide (best efficacy/tolerability), valproate, lamotrigine | Ethosuximide does not work for GTC or focal seizures |
| Myoclonic | Valproate; + levetiracetam, zonisamide, clonazepam, or clobazam if refractory | |
| Mixed IGE (GTC + absence/myoclonic) | Valproate | Levetiracetam increasingly preferred for women of childbearing age given valproate teratogenicity |
| Atonic/Lennox-Gastaut | Valproate, lamotrigine, topiramate, rufinamide |
Narrow-spectrum drugs (carbamazepine, phenytoin, gabapentin, pregabalin, tiagabine, oxcarbazepine) are appropriate only for focal seizures — do not use for generalized epilepsy.
Phenobarbital
Historically first-line but now reserved for situations where no alternatives exist, due to sedation in adults, hyperactivity in children, and cognitive effects.
3. Initiating & Adjusting Therapy
- Start low, titrate slowly — wait ≥5 half-lives between dose increases (steady state)
- If seizures persist at maximum tolerated dose → add a second drug, then taper the first
- Switch drugs one at a time; never abruptly discontinue the first ASM
- Polypharmacy: combining agents with different mechanisms of action is theoretically optimal; typical combinations: carbamazepine or lamotrigine + levetiracetam or valproate
4. When to Stop Therapy
Candidates for discontinuation after full seizure control must meet all of:
- Seizure-free for 1–5 years
- Single seizure type (generalized better than focal)
- Normal neurological exam and intelligence
- No family history of epilepsy
- Normal EEG
Taper gradually over 2–3 months. Most recurrences occur within the first 3 months. Advise against driving or swimming during the taper/withdrawal period.
5. Refractory Epilepsy (~30% of patients)
When ≥2 ASMs fail, epilepsy is considered drug-resistant. Options include:
A. Further pharmacotherapy
- Rational polypharmacy with drugs of different mechanisms
- Add cenobamate, zonisamide, brivaracetam, topiramate, or lacosamide
- For refractory absence: valproate + ethosuximide combination
- For refractory myoclonic: add levetiracetam, zonisamide, or clonazepam
B. Epilepsy Surgery
- Resective surgery (most commonly anterior temporal lobectomy for mesial TLE): 80–90% seizure freedom in well-selected candidates
- Presurgical evaluation: video-EEG, high-resolution MRI, neuropsychological testing, and optionally ictal SPECT/PET
C. Vagus Nerve Stimulation (VNS)
- For patients who are not surgical candidates or fail surgery
- ~50% of patients achieve ≥50% reduction in seizure frequency
D. Deep Brain Stimulation (DBS)
- Anterior nucleus of thalamus (SANTE trial): 68% of patients had >50% seizure reduction at 5 years
- FDA-approved for drug-resistant epilepsy
E. Responsive Neurostimulation (RNS / NeuroPace)
- Detects ictal activity and delivers targeted stimulation
- ~48–70% seizure reduction in long-term follow-up; FDA-approved
- Particularly valuable for mesial temporal lobe epilepsy (70% median seizure reduction at 6 years)
F. Ketogenic Diet
- High-fat, low-carbohydrate diet; evidence-based especially in children
- Mechanism involves altered brain metabolism reducing seizure threshold
6. Status Epilepticus
- First-line: IV lorazepam (0.1 mg/kg, max 4 mg)
- Second-line: IV phenytoin / fosphenytoin or IV valproate or IV phenobarbital
- Urgent in nature — requires ICU management if refractory
7. Special Populations
Women of childbearing age:
- Avoid valproate if possible — significant teratogen (neural tube defects, cognitive effects)
- Prefer levetiracetam or lamotrigine
- Pregnancy registries document risks of individual ASMs
Febrile seizures:
- Treatment generally not required
- Diazepam every 8 hours can be used for prophylaxis if carefully selected
Recent Evidence (PubMed 2024–2025)
- Li C et al. Int J Mol Sci 2025 — Reviews mechanisms of AED combinations and future strategies, highlighting that rational polypharmacy targeting different pathways (Na⁺ channels, GABA, AMPA, synaptic vesicle protein SV2A) improves outcomes (PMID 40362274)
- Auvin S. Rev Neurol 2025 — Targeted therapies in epilepsy: genetic testing now enables precision medicine (e.g., cannabidiol in Dravet syndrome, mTOR inhibitors in TSC, quinidine in KCNT1 mutations) (PMID 40221358)
- Perucca P et al. Epilepsia 2025 — Updated pregnancy registry data refining teratogenicity estimates by ASM exposure (PMID 39540312)
⚠️ The 2025 targeted therapy review (PMID 40221358) is notable: genetic/precision medicine is now an emerging treatment layer — genetic testing before drug selection is increasingly recommended, especially in children.
- Harrison's Principles of Internal Medicine 22E (2025); Bradley & Daroff's Neurology in Clinical Practice; Swanson's Family Medicine Review
pathophysiology
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Searching Images
epilepsy pathophysiology seizure mechanism excitatory inhibitory
I now have comprehensive material. Let me compile the full pathophysiology response.
Epilepsy: Pathophysiology & Mechanisms
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics; Bradley & Daroff's Neurology in Clinical Practice; Kaplan & Sadock's Comprehensive Textbook of Psychiatry; Lippincott's Illustrated Pharmacology; Basic Medical Biochemistry
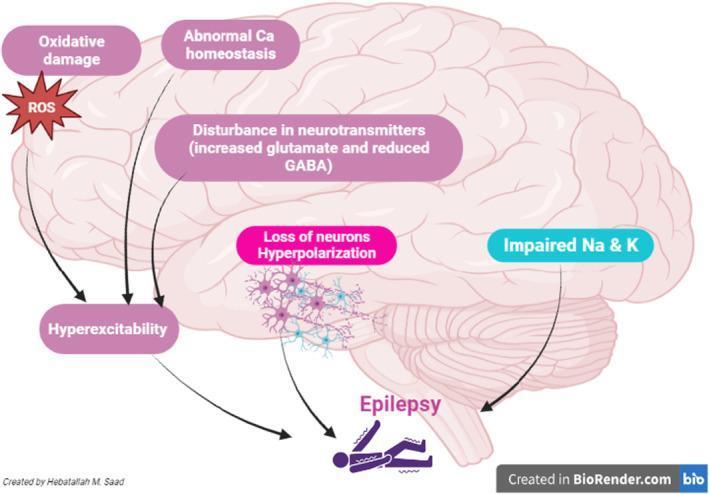
Core Concept: Failure of Excitation/Inhibition Balance
At its foundation, a seizure results from abnormal, synchronous, high-frequency neuronal firing — a state produced when excitatory drive overwhelms inhibitory control. The key neurotransmitters involved are:
- Glutamate — principal excitatory neurotransmitter (acts at NMDA, AMPA, kainate receptors)
- GABA — principal inhibitory neurotransmitter (acts at GABA-A and GABA-B receptors)
Pharmacological evidence is compelling: GABA-A receptor antagonists and glutamate receptor agonists reliably provoke seizures in experimental models; conversely, GABA enhancers and glutamate antagonists suppress them.
1. Neuronal Mechanisms: The Depolarization Shift
The cellular hallmark of epileptic activity is the depolarization shift (DS) — the intracellular correlate of the interictal spike seen on EEG:
- A large sustained membrane depolarization drives a burst of high-frequency action potentials
- Generated by a large excitatory synaptic current, amplified by voltage-gated intrinsic membrane currents (Na⁺, Ca²⁺, K⁺ channels)
- In physiological firing, Na⁺ channels open → action potential → spontaneous inactivation → refractory period → recovery
- In epileptic firing, high-frequency repetitive action potentials are sustained because channels rapidly recover from inactivation, bypassing the refractory brake
- Drugs like carbamazepine, phenytoin, and lamotrigine exploit this by prolonging Na⁺ channel inactivation, selectively suppressing high-frequency pathological firing without affecting normal slow firing
2. Synaptic Mechanisms
A. Reduced GABAergic Inhibition
- GABA-A receptor activation opens Cl⁻ channels → Cl⁻ influx → membrane hyperpolarization → ↓ neuronal excitability
- Reduced GABA synthesis, increased GABA catabolism, or GABA-A receptor dysfunction → disinhibition → seizure threshold falls
- Penicillin-induced seizures are a classic experimental model of GABA-A blockade
- Benzodiazepines and barbiturates both enhance GABA-A receptor-mediated Cl⁻ conductance (through distinct binding sites), which underlies their antiseizure efficacy
B. Enhanced Glutamatergic Excitation
- Excess activation at NMDA receptors (kainic acid model), AMPA receptors, or kainate receptors triggers seizures
- NMDA receptors allow Ca²⁺ influx, which further amplifies neuronal excitability and triggers downstream signaling cascades
- Abnormal Ca²⁺ homeostasis contributes to both acute seizure generation and chronic neuronal damage
C. Ion Channel Roles
| Channel | Role in Seizures |
|---|---|
| Na⁺ channels | Persistent/rapid recovery from inactivation → sustained depolarization |
| T-type Ca²⁺ channels | Low-threshold Ca²⁺ currents → thalamo-cortical spike-wave oscillations (absence seizures) |
| K⁺ channels (KCNQ) | Loss of function → reduced repolarization → hyperexcitability |
| HCN channels | Abnormal Ih current → altered oscillatory behavior |
3. Network Mechanisms: Synchronization & Spread
A seizure is not just a single-neuron event — it requires abnormal synchronization across neuronal networks:
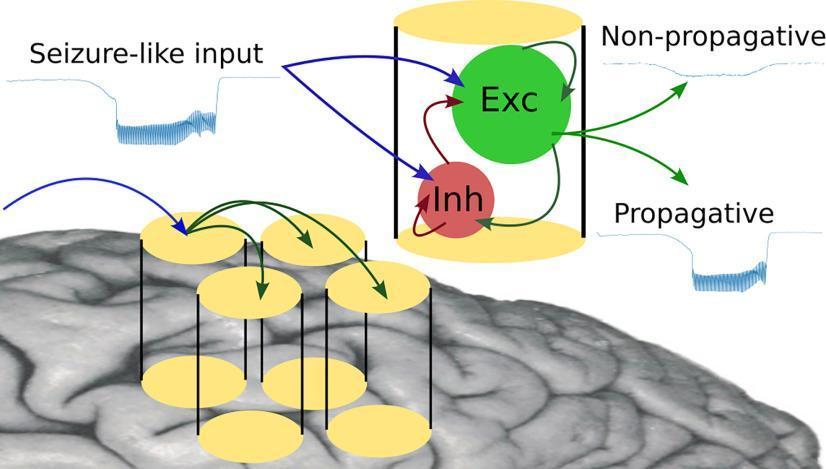
- In focal epilepsy, a localized seizure focus (irritative zone) initiates the discharge; surrounding tissue is initially inhibited (penumbra of hypometabolism)
- The seizure can remain focal or spread to adjacent and distant structures depending on the integrity of inhibitory networks
- In generalized epilepsy, the thalamo-cortical loop is central: rhythmic 3-Hz spike-and-wave discharges of absence epilepsy arise from cortico-thalamic oscillations involving thalamic relay neurons and reticular nucleus GABAergic interneurons
- Loss of inhibitory interneuron function (e.g., parvalbumin-positive fast-spiking interneurons) is a particularly critical failure point — these cells normally provide powerful, fast inhibition to pyramidal neurons
4. Epileptogenesis: How a Normal Brain Becomes Epileptic
Epileptogenesis refers to the process by which a brain is transformed from normal to chronically epileptic, often after an initial insult:
Kindling Model
- Repeated subthreshold electrical stimulations of limbic structures (e.g., amygdala) → progressive intensification of seizure response → eventually spontaneous seizures persisting for life
- Demonstrates that repeated seizure activity itself modifies neural circuits — seizures beget seizures
Status Epilepticus → Hippocampal Sclerosis
- Prolonged seizures (status epilepticus) cause hippocampal neuronal death (especially CA1 and CA3 pyramidal cells, hilar interneurons)
- This mirrors the clinical observation that complicated febrile seizures in young children precede the development of mesial temporal lobe epilepsy years later
- The latent period between the initial insult and the onset of spontaneous seizures involves extensive network reorganization
Mossy Fiber Sprouting
- Surviving dentate granule cells (DGCs) sprout new axons (mossy fibers) that form aberrant recurrent excitatory connections within the dentate gyrus
- This creates a positive feedback loop that amplifies excitability in the hippocampal circuit
- Normally, DGCs are relatively non-excitable ("sparse activation"), acting as a gate against seizure propagation from entorhinal cortex to hippocampus — mossy fiber sprouting breaks this gate
5. Genetic Mechanisms
Many epilepsy syndromes involve channelopathies — mutations in genes encoding ion channels or their regulatory subunits:
| Gene | Channel | Syndrome |
|---|---|---|
| SCN1A | Na⁺ channel α-subunit (Nav1.1) | Dravet syndrome — mutations impair interneuron Na⁺ channel function → disinhibition; ~85% of Dravet patients carry SCN1A mutations |
| GABRA1, GABRG2 | GABA-A receptor subunits | Childhood absence epilepsy (CAE) — mutations reduce surface expression of GABA-A receptors via mRNA degradation |
| GABRB3 | GABA-A receptor β3 subunit | CAE — alters ion channel function without affecting receptor trafficking |
| KCNQ2/3 | M-type K⁺ channels | Benign neonatal epilepsy — reduced repolarizing current |
| SCN2A | Nav1.2 | Infantile epileptic encephalopathy |
Key insight for Dravet syndrome: SCN1A mutations preferentially reduce Na⁺ channel function in GABAergic interneurons (not pyramidal cells), causing disinhibition of excitatory circuits — paradoxically, sodium channel loss-of-function produces a gain of excitability at the network level.
6. Drug Mechanism Summary
| Mechanism | Drugs |
|---|---|
| Na⁺ channel fast inactivation ↑ | Phenytoin, carbamazepine, lamotrigine, oxcarbazepine, valproate, eslicarbazepine |
| Na⁺ channel slow inactivation ↑ | Lacosamide |
| T-type Ca²⁺ channel block | Ethosuximide, valproate (→ absence seizures) |
| GABA-A allosteric enhancement | Benzodiazepines, barbiturates, clobazam, stiripentol |
| GABA reuptake inhibition | Tiagabine |
| GABA-transaminase inhibition | Vigabatrin (↑ synaptic GABA) |
| SV2A (synaptic vesicle protein) binding | Levetiracetam, brivaracetam (↓ neurotransmitter release) |
| AMPA receptor antagonism | Perampanel, topiramate |
| α2δ Ca²⁺ channel modulation | Gabapentin, pregabalin |
| Multiple mechanisms | Valproate, topiramate, zonisamide |
Summary
Seizures arise when neuronal excitability exceeds inhibitory control through a combination of: enhanced glutamatergic transmission, impaired GABAergic inhibition, and aberrant ion channel function. Epileptogenesis — the transformation to a chronically epileptic state — involves structural changes including hippocampal sclerosis, mossy fiber sprouting, and interneuron loss. Genetic epilepsies most commonly involve channelopathies that preferentially impair inhibitory interneurons, triggering network disinhibition. All currently available ASMs act symptomatically (suppressing seizures) rather than antiepilelptogenically (preventing the underlying process).
— Goodman & Gilman's Pharmacological Basis of Therapeutics; Bradley & Daroff's Neurology in Clinical Practice; Kaplan & Sadock's Comprehensive Textbook of Psychiatry
This is a shared conversation. Sign in to Orris to start your own chat.