Polycythemia
polycythemia vera management
Polycythemia
Definition
Classification
| Type | Mechanism | Examples |
|---|---|---|
| Relative | Normal red cell mass, decreased plasma volume (hemoconcentration) | Dehydration, vomiting, diarrhea, burns, diuretic therapy, Gaisböck syndrome |
| Absolute - Primary | Intrinsic abnormality of hematopoietic precursors; low/normal EPO | Polycythemia vera, familial EPO receptor mutations |
| Absolute - Secondary (compensatory) | Appropriate elevated EPO response to hypoxia | High altitude, lung disease, cyanotic heart disease, carboxyhemoglobinemia |
| Absolute - Secondary (inappropriate) | Autonomous EPO secretion; normoxic tissues | Renal cell carcinoma, hepatocellular carcinoma, cerebellar hemangioblastoma, Wilms tumor |
| Genetic/Congenital | HIF pathway mutations or EPO receptor mutations | Chuvash polycythemia (VHL mutation), prolyl hydroxylase mutations, primary familial congenital polycythemia |
Spurious polycythemia (Gaisböck syndrome): Red cell mass is often high-normal, plasma volume is low-normal. Seen predominantly in obese, hypertensive men who smoke.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Table 14.8
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Box 33.4
Polycythemia Vera (PV)
Pathogenesis
- JAK2 V617F (exon 14): Found in >95% of PV cases. This point mutation in the pseudokinase domain (JH2) removes the auto-inhibitory function of JAK2, constitutively activating JAK-STAT signaling independent of EPO.
- JAK2 exon 12 mutations: Found in JAK2 V617F-negative PV patients.
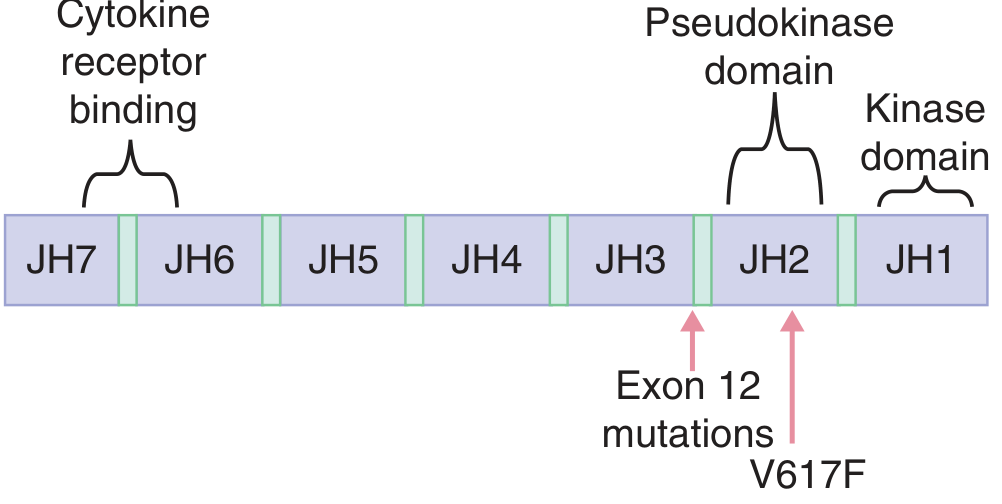
Morphology / Pathology
-
Bone marrow: Hypercellular - increased erythroid, myeloid, and megakaryocytic forms (panmyelosis); 10% have marrow fibrosis at diagnosis
-
Spleen: Mildly enlarged (250-300 g) from vascular congestion; foci of extramedullary hematopoiesis
-
Liver: Enlarged with foci of extramedullary hematopoiesis
-
Thromboses and infarctions: Common in heart, spleen, and kidneys (due to hyperviscosity and vascular stasis)
-
Hemorrhages: Occur in ~one-third of patients (GI tract, oropharynx, brain)
-
Peripheral blood: Basophilia is characteristic; dysfunctional giant platelets; megakaryocyte fragments
-
Robbins & Kumar Basic Pathology, p. 407
Clinical Features
| Symptom/Sign | Mechanism |
|---|---|
| Plethora (ruddy complexion) | Increased blood volume + sluggish flow in skin capillaries + deoxygenation |
| Cyanosis | Slow capillary transit -> excess deoxygenated Hb in subpapillary venous plexus |
| Pruritus (especially after hot bath - "aquagenic pruritus") | Histamine release from neoplastic basophils |
| Headache, dizziness | Hyperviscosity, reduced cerebral blood flow |
| Hypertension (~1/3 of patients) | Increased blood viscosity -> increased peripheral resistance |
| Thrombosis (~30%) | Hyperviscosity + vascular stasis; affects brain, heart, hepatic vein (Budd-Chiari) |
| Hemorrhage (5-10% life-threatening) | Dysfunctional platelets; minor epistaxis and gum bleeding are common |
| Erythromelalgia | Burning pain + erythema/warmth of hands and feet; microvascular disturbances |
| Peptic ulcer | Histamine-stimulated gastric acid secretion |
| Gout (5-10%) | High cell turnover -> hyperuricemia |
| Splenomegaly | Extramedullary hematopoiesis |
Laboratory Findings
| Parameter | Finding |
|---|---|
| RBC count | 6-10 million/µL (normal ~4.5-5.5) |
| Hematocrit | Often 60-70% (normal 40-45%) |
| Hemoglobin | Median 18.4 g/dL (large series) |
| WBC | Up to 50,000/µL; basophilia present |
| Platelets | Often >400,000/µL; functionally abnormal |
| Serum EPO | Low (key distinguishing feature from secondary polycythemia) |
| JAK2 V617F | Positive in >95% |
WHO Diagnostic Criteria (2016/2022)
- Hb >16.5 g/dL (men) or >16.0 g/dL (women), OR Hct >49% (men) or >48% (women), OR increased red cell mass
- Bone marrow biopsy: hypercellular (panmyelosis) with pleomorphic mature megakaryocytes
- Presence of JAK2 V617F or JAK2 exon 12 mutation
- Subnormal serum erythropoietin level
Natural History and Complications
- Thrombotic/hemorrhagic complications: The main causes of morbidity and mortality
- Spent phase (post-PV myelofibrosis): 15-20% of cases after ~10 years; bone marrow fibrosis, massive splenomegaly, worsening cytopenias. JAK2 inhibitors (ruxolitinib) are approved for this phase.
- Blast crisis / AML transformation: Less common than in CML; more rare overall
Treatment
| Intervention | Rationale |
|---|---|
| Phlebotomy | Reduces red cell mass; mainstay - median survival increases to ~10 years |
| Low-dose aspirin | Reduces microvascular and thrombotic risk |
| Hydroxyurea | Cytoreduction in high-risk patients |
| Ruxolitinib (JAK2 inhibitor) | Approved for PV refractory to hydroxyurea; also used in spent-phase PV myelofibrosis |
| Ropeginterferon alfa-2b | IFN-based therapy; recent meta-analysis (PMID 41238945) shows efficacy and safety in PV |
Secondary Polycythemia - Key Points
- EPO is elevated (unlike PV where it is suppressed)
- Physiologic/compensatory: High altitude (RBC count rises to 6-7 million/mm³), chronic lung disease, cyanotic congenital heart disease - all due to tissue hypoxia stimulating renal EPO production
- Paraneoplastic: Renal cell carcinoma, hepatocellular carcinoma, cerebellar hemangioblastoma produce EPO autonomously
- HIF pathway mutations (Chuvash polycythemia - homozygous VHL mutation; prolyl hydroxylase mutations) stabilize HIF-1α, which constitutively upregulates EPO transcription even at normal oxygen tension
- Rarely, high-affinity hemoglobin mutants (impaired O2 unloading) or 2,3-BPG deficiency cause secondary polycythemia
Measurement of Red Cell Mass
- Red cell volume: ⁵¹Cr-labeled autologous RBCs
- Plasma volume: ¹²⁵I-labeled albumin
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Guyton and Hall Textbook of Medical Physiology
If it's due to smoking
Polycythemia Due to Smoking
Mechanism 1: Absolute Secondary Polycythemia - "Smoker's Polycythemia"
Pathophysiology
- Reduces functional O2-carrying capacity - the hemoglobin bound to CO cannot carry oxygen
- Shifts the oxygen-hemoglobin dissociation curve to the left - even the remaining oxyhemoglobin releases O2 less readily to tissues
- Together, these produce functional tissue hypoxia despite a normal arterial PO2
- The kidneys sense reduced O2 delivery and respond by increasing EPO secretion
- Elevated EPO drives compensatory erythroid proliferation in the marrow - true absolute polycythemia
Key point: O2 saturation measured by standard pulse oximetry is falsely normal in smokers because it cannot distinguish COHb from oxyhemoglobin. Arterial blood gas with co-oximetry is needed to measure true COHb levels.
Mechanism 2: Relative Polycythemia - Gaisböck Syndrome
- Red cell mass is high-normal (not truly elevated)
- Plasma volume is reduced (hemoconcentration)
- The net result is an elevated hematocrit without a true increase in total circulating RBCs
- Henry's Clinical Diagnosis and Management by Laboratory Methods
Summary: How Smoking Causes Both Types
| Type | Mechanism | EPO | Red Cell Mass | COHb |
|---|---|---|---|---|
| Smoker's polycythemia (absolute secondary) | CO -> functional hypoxia -> elevated EPO | High | Truly elevated | High |
| Gaisböck syndrome (relative) | Plasma volume contraction | Normal | Normal to high-normal | Normal/mildly elevated |
Diagnostic Approach (Harrison's)
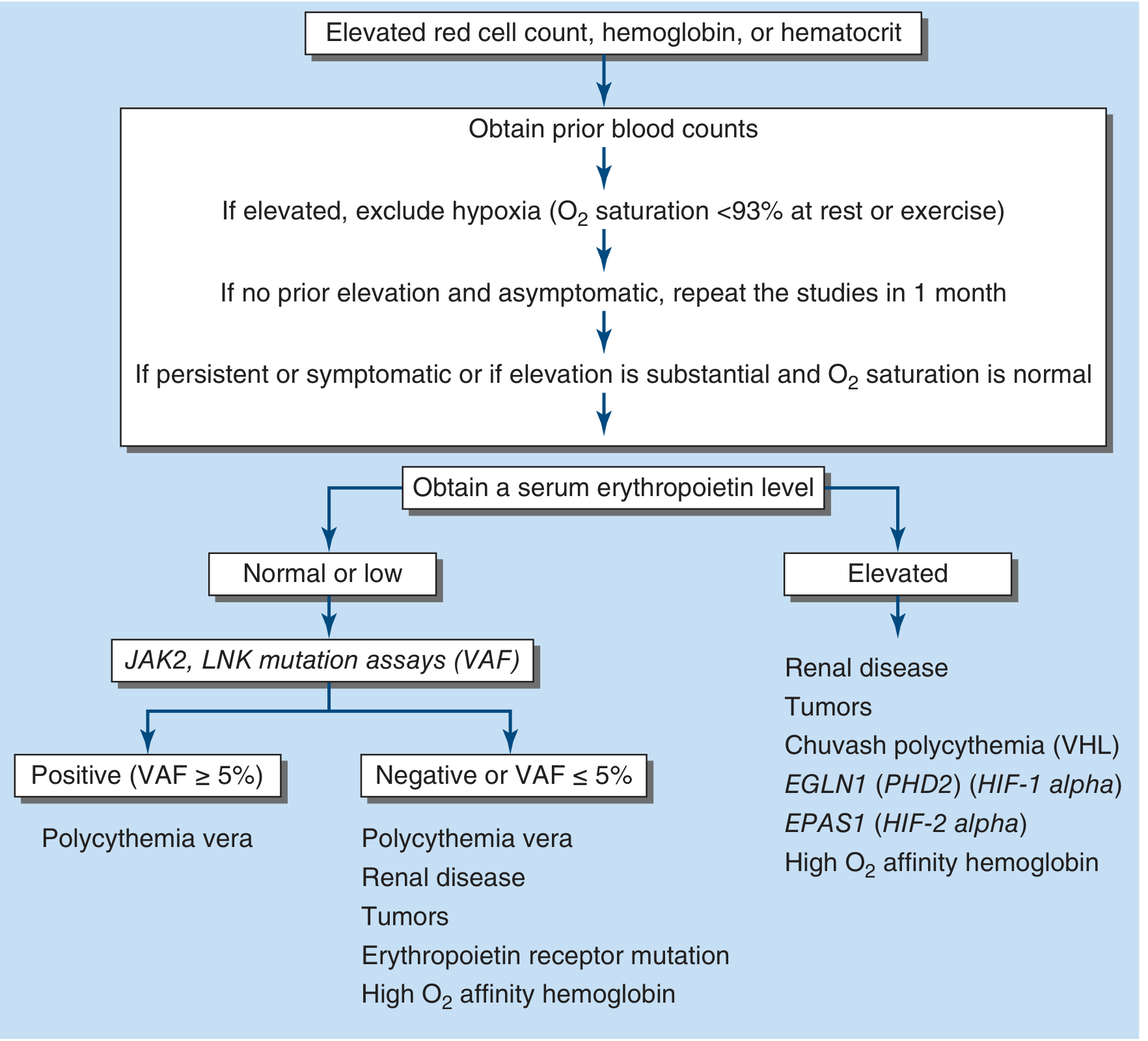
- Check O2 saturation - may be falsely normal in smokers by pulse oximetry
- Measure serum EPO - will be elevated in smoker's polycythemia (unlike PV where it is low)
- If EPO is elevated and O2 saturation appears normal, measure COHb by co-oximetry
- High COHb + elevated EPO + smoking history = "Smoker's polycythemia" - diagnosis confirmed
Management
| Approach | Details |
|---|---|
| Smoking cessation | First-line and definitive - COHb normalizes within hours to days of stopping; polycythemia resolves over weeks |
| Phlebotomy | For those unable to quit, to reduce hematocrit and hyperviscosity risk |
| Aspirin | Consider for thrombotic risk reduction |
"Such patients should be urged to stop smoking. Those who cannot stop smoking require phlebotomy to control their polycythemia." - Harrison's Principles of Internal Medicine, 22e
Why Smoking Polycythemia Matters Clinically
- The elevated hematocrit increases blood viscosity -> thrombotic risk (stroke, MI, DVT)
- It can mimic polycythemia vera on initial labs - the key distinguishing feature is elevated (not low) serum EPO in smoker's polycythemia
- Standard pulse oximetry will miss the underlying hypoxia - you must use co-oximetry to detect COHb
How do you distinguish smoker's polycythemia from PV using labs?
Distinguishing Smoker's Polycythemia from Polycythemia Vera: Lab Comparison
The Core Logic
- In PV: the bone marrow proliferates autonomously, EPO is suppressed, all cell lines are involved
- In smoker's polycythemia: the marrow responds normally to a CO-driven hypoxic signal; only red cells increase
Side-by-Side Lab Comparison
| Test | Smoker's Polycythemia | Polycythemia Vera |
|---|---|---|
| Serum EPO | Elevated (appropriate response to functional hypoxia) | Low / subnormal (suppressed by autonomous erythropoiesis) - this is a WHO minor criterion |
| JAK2 V617F / exon 12 mutation | Absent | Present in >95% (major WHO criterion) |
| Carboxyhemoglobin (COHb) - co-oximetry | Elevated (>3-5% in smokers, can be >10-15% in heavy smokers) | Normal |
| Pulse oximetry (SpO2) | Falsely normal - cannot detect COHb | Normal |
| True arterial O2 saturation (ABG + co-oximetry) | Reduced (functional hypoxia revealed) | Normal (>92%) |
| WBC / Leukocyte count | Normal (EPO stimulates only erythroid lineage) | Elevated - leukocytosis common; up to 50,000/µL |
| Platelet count | Normal | Elevated (>400,000/µL), often dysfunctional |
| Basophilia | Absent | Present - characteristic of PV and other MPNs |
| Splenomegaly | Absent | Present (extramedullary haematopoiesis, vascular congestion) |
| Bone marrow biopsy | Normal cellularity | Hypercellular - panmyelosis with pleomorphic megakaryocytes (major WHO criterion) |
| Serum uric acid | Normal | Elevated (high cell turnover; gout in 5-10%) |
| LDH | Normal | Often elevated |
| Red cell mass (⁵¹Cr) | Truly elevated | Truly elevated |
The Decisive Tests - In Order of Priority
Step 1: Serum EPO
- Low EPO -> strongly suggests PV -> proceed to JAK2 testing
- Elevated EPO -> suggests secondary polycythemia (smoking, hypoxia, tumour) -> check COHb
Step 2: JAK2 V617F mutation assay
- Positive (VAF ≥5%) -> PV confirmed (no further workup needed if other criteria met)
- Negative -> consider JAK2 exon 12, renal disease, tumours, or other causes
Step 3: COHb by co-oximetry (arterial blood gas)
- Elevated COHb + smoking history + elevated EPO = smoker's polycythemia
- Do not rely on pulse oximetry alone - it reads COHb as oxyhaemoglobin and gives a falsely normal SpO2
Step 4: CBC differential
- Leukocytosis + thrombocytosis + basophilia accompany the erythrocytosis
- In smoker's polycythemia, WBC and platelets are normal - EPO only drives the erythroid lineage
"As erythropoietin stimulates only the red cell pathway, WBC and platelet counts should be unchanged [in secondary polycythemia]." - Rosen's Emergency Medicine
WHO Diagnostic Criteria for PV (2016) - for Reference
- Hb >16.5 g/dL (men) or >16.0 g/dL (women), OR Hct >49%/48%, OR elevated red cell mass
- Bone marrow: trilineage proliferation with pleomorphic mature megakaryocytes
- JAK2 V617F or exon 12 mutation
- Subnormal serum EPO
Quick Summary Table
| Feature | Smoker's Poly | PV |
|---|---|---|
| EPO | ↑ | ↓ |
| JAK2 | Negative | Positive (>95%) |
| COHb | ↑ | Normal |
| WBC/Platelets | Normal | ↑ |
| Basophilia | No | Yes |
| Splenomegaly | No | Yes |
| Bone marrow | Normal | Panmyelosis |
| Resolves with smoking cessation | Yes | No |
- Harrison's Principles of Internal Medicine, 22e
- Rosen's Emergency Medicine, WHO Box 109.16
- Robbins & Kumar Basic Pathology
How to approach case of polycythemia
Approach to a Case of Polycythemia
When to Suspect It
- Hb >17 g/dL (men) or >15 g/dL (women)
- Hct >50% (men) or >45% (women)
- Hct >60% (men) or >55% (women) is almost invariably a true elevated red cell mass
Step 1: History and Physical Examination
| Ask About | Significance |
|---|---|
| Smoking | CO-driven secondary polycythemia (smoker's polycythemia) |
| High altitude living | Physiologic/compensatory secondary polycythemia |
| Chronic lung disease, snoring/sleep apnea | Hypoxic drive; secondary polycythemia |
| Cyanotic congenital heart disease | Right-to-left shunt causing hypoxia |
| Diuretic use, vomiting, diarrhoea, burns | Plasma volume contraction -> relative polycythemia |
| Testosterone, EPO, SGLT2 inhibitor (gliflozin) use | Exogenous causes of erythrocytosis |
| Aquagenic pruritus | Highly specific for PV |
| Headache, dizziness, visual changes, tinnitus | Hyperviscosity symptoms |
| Thrombotic events (DVT, stroke, MI, Budd-Chiari) | Complication of PV or any polycythemia |
| Family history | Congenital causes (EPO receptor mutation, Chuvash polycythemia) |
- Ruddy (plethoric) complexion - hallmark of polycythemia
- Splenomegaly - strongly favours PV (extramedullary haematopoiesis)
- Cyanosis - suggests right-to-left shunt or severe hypoxia
- Signs of chronic lung disease (clubbing, barrel chest, wheeze)
- Hypertension - common in both PV and Gaisböck syndrome
Step 2: Confirm True (Absolute) vs. Relative Polycythemia
| Indicator | Relative Polycythemia | Absolute Polycythemia |
|---|---|---|
| Red cell mass (⁵¹Cr) | Normal (<36 mL/kg men, <32 mL/kg women) | Elevated (>36/32 mL/kg) |
| Clinical context | Dehydration, diuretics, burns, Gaisböck syndrome | Persistent on repeat testing |
| WBC/Platelets | Normal | May be elevated (PV) |
In practice, ⁵¹Cr red cell mass measurement is rarely performed now. Instead, confirm persistence on repeat CBC after adequate hydration, and proceed to EPO measurement if Hb/Hct remains elevated.
- Acute dehydration (vomiting, diarrhoea, burns)
- Diuretic therapy
- Gaisböck (spurious/stress) polycythemia - obese, hypertensive, smoking men with chronically contracted plasma volume
Step 3: Check O₂ Saturation First
- If O₂ sat <92-93% -> patient is hypoxic -> secondary polycythemia likely -> investigate for heart/lung disease or high altitude
- If O₂ sat is normal (≥93%) -> proceed to EPO measurement
Caveat for smokers: Standard pulse oximetry is unreliable - it cannot distinguish COHb from oxyHb and will read falsely normal. Use ABG with co-oximetry to get the true O₂ saturation and measure COHb directly.
Step 4: Measure Serum Erythropoietin (EPO) - The Pivotal Test
If EPO is LOW or Undetectable
- Test for JAK2 V617F mutation (and exon 12 if V617F negative)
- JAK2 VAF ≥5% -> Polycythemia Vera confirmed
- JAK2 negative -> still possible PV (check exon 12), or rare EPO receptor mutation
- Abdominal ultrasound - assess spleen size (splenomegaly supports PV)
- Supporting labs for PV: leukocytosis, thrombocytosis, absolute basophilia, elevated LDH, elevated uric acid
- Bone marrow biopsy (if needed to complete WHO criteria): trilineage hyperplasia (panmyelosis), pleomorphic megakaryocytes
If EPO is ELEVATED
Step 5: Elevated EPO - Narrow the Cause
Elevated EPO
|
├── O₂ sat LOW (<92%)
│ └── Lung disease, cyanotic heart disease, high altitude,
│ sleep apnoea, right-to-left shunt
│ -> Treat/investigate the underlying condition
│
└── O₂ sat NORMAL (≥93%)
|
├── SMOKER? -> Check COHb (co-oximetry on ABG)
│ COHb elevated -> "Smoker's polycythemia"
│ -> Smoking cessation; phlebotomy if can't quit
│
├── High O₂ affinity haemoglobin?
│ -> P50 assay (reduced), haemoglobin electrophoresis
│
└── Autonomous EPO secretion (tumour/renal)?
-> CT abdomen/pelvis
-> Renal cell carcinoma, hepatocellular carcinoma,
Wilms tumour, cerebellar haemangioblastoma,
uterine leiomyoma, renal cysts
-> MRI brain/cerebellum if neurologic signs
Step 6: Complete the Workup Based on the Branch
| Diagnosis | Confirmatory Tests |
|---|---|
| Polycythemia Vera | JAK2 V617F/exon 12; bone marrow biopsy; subnormal EPO |
| Smoker's polycythemia | COHb elevated on co-oximetry; elevated EPO; resolves with cessation |
| Hypoxic secondary | ABG (low PaO₂/SpO₂); PFTs; echo (shunt); sleep study |
| Paraneoplastic | CT abdomen/pelvis; LFTs; renal function; tumour markers |
| High-affinity Hb | P50 (low), haemoglobin electrophoresis, DNA analysis |
| Chuvash/VHL polycythemia | VHL gene mutation; normal O₂ sat; elevated EPO; family history |
| Relative/Gaisböck | Normalises with rehydration; red cell mass normal |
Summary Algorithm
Elevated Hb/Hct on CBC
|
v
[1] HISTORY + EXAM
(smoking, altitude, drugs, aquagenic pruritus, splenomegaly)
|
v
[2] REPEAT CBC AFTER HYDRATION
-> If normalises: Relative polycythemia - manage cause
-> If persists: True absolute polycythemia
|
v
[3] CHECK O₂ SATURATION (ABG + co-oximetry, NOT just pulse ox)
-> <92%: Hypoxic secondary polycythemia
-> ≥93%: Proceed to EPO
|
v
[4] SERUM EPO LEVEL <--- THE PIVOTAL TEST
/ \
LOW HIGH
(PV likely) (Secondary)
| |
v v
[5] JAK2 V617F O₂ sat normal?
+ exon 12 | |
Abdo U/S Smoker? No
BM biopsy COHb↑ -> CT abdomen (tumour)
-> Smoker's Poly P50 (high-affinity Hb)
VHL gene (Chuvash)
Red Flags Requiring Urgent Action
| Feature | Significance |
|---|---|
| Hct >60% | Exponential rise in blood viscosity -> urgent phlebotomy |
| Neurologic symptoms (stroke, TIA, visual loss) | Hyperviscosity thrombosis |
| Budd-Chiari syndrome | Hepatic vein thrombosis - classic PV complication |
| Bleeding + markedly elevated platelets | Paradoxical bleeding from platelet dysfunction in PV |
- Harrison's Principles of Internal Medicine 22e, pp. 494-495
- Rosen's Emergency Medicine, Box 109.15-109.16
- Tintinalli's Emergency Medicine
- Murray & Nadel's Textbook of Respiratory Medicine
Create a clinical decision tree poster for polycythemia workup
Clinical decision tree poster for polycythemia workup. Medical education style, clean white background, professional dark navy and teal color scheme with red accent boxes for diagnoses. Title at top: "APPROACH TO POLYCYTHEMIA" with subtitle "Clinical Decision Tree" The flowchart flows top to bottom with the following nodes connected by arrows: START BOX (dark navy): "Elevated Hb, Hct, or RBC on CBC Men: Hb >17 g/dL or Hct >50% Women: Hb >15 g/dL or Hct >45%" Arrow down to: STEP 1 BOX (teal): "HISTORY & PHYSICAL • Smoking, altitude, lung disease, sleep apnea • Diuretics, testosterone, EPO, SGLT2 inhibitors • Aquagenic pruritus → PV • Splenomegaly → PV • Plethora, hypertension, thrombosis history" Arrow down to: STEP 2 BOX (teal): "REPEAT CBC AFTER HYDRATION" Two branches: - Left branch (grey box): "Normalises → RELATIVE POLYCYTHEMIA Causes: Dehydration, diuretics, burns Gaisböck syndrome (obese, HTN, smoker)" - Right branch continues down: "Persists → ABSOLUTE POLYCYTHEMIA" Arrow down to: STEP 3 BOX (teal): "ARTERIAL BLOOD GAS + CO-OXIMETRY (NOT pulse oximetry alone in smokers)" Two branches: - Left (orange box): "SpO₂ <92% HYPOXIC SECONDARY POLYCYTHEMIA → PFTs, Echo, Sleep study Causes: COPD, cyanotic heart disease, high altitude, sleep apnea" - Right continues: "SpO₂ ≥93% → Proceed" Arrow down to: STEP 4 BOX (dark navy, larger): "⭐ SERUM ERYTHROPOIETIN LEVEL THE PIVOTAL TEST" Two large branches LEFT and RIGHT: LEFT BRANCH - LOW/SUPPRESSED EPO: Box (red): "PRIMARY POLYCYTHEMIA VERA → JAK2 V617F mutation assay → JAK2 exon 12 if V617F negative → Abdominal ultrasound (splenomegaly) → Bone marrow biopsy Supporting: Leukocytosis, thrombocytosis, basophilia WHO: 3 major OR 2 major + minor criterion" RIGHT BRANCH - ELEVATED EPO: Three sub-branches: 1. Box (orange): "SMOKER with normal SpO₂ → COHb on co-oximetry elevated SMOKER'S POLYCYTHEMIA Rx: Smoking cessation / Phlebotomy" 2. Box (orange): "NON-SMOKER, normal SpO₂ → CT abdomen/pelvis PARANEOPLASTIC Renal cell carcinoma, Hepatocellular Ca, Cerebellar hemangioblastoma, Wilms tumor, Uterine leiomyoma" 3. Box (orange): "High O₂ affinity Hb / Genetic → P50 assay, Hb electrophoresis → VHL/EGLN1 gene testing Chuvash polycythemia, High-affinity Hb" Bottom section - RED ALERTS BOX (red border): "🚨 URGENT: Hct >60% → Phlebotomy immediately Neurologic symptoms → Hyperviscosity thrombosis Budd-Chiari syndrome → Classic PV complication Bleeding + high platelets → Platelet dysfunction in PV" Footer: "Based on Harrison's Internal Medicine 22e | WHO 2016 Criteria"
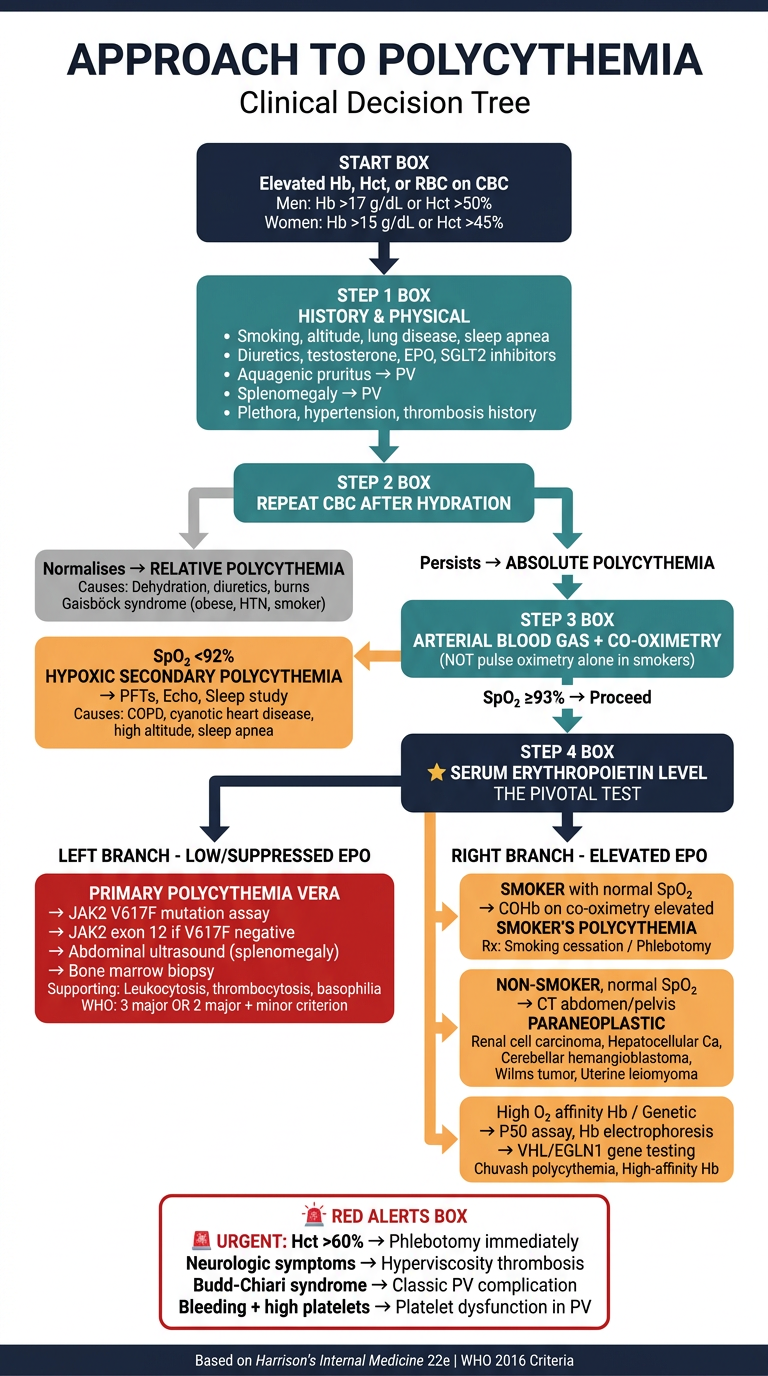
- Trigger - Hb/Hct thresholds that prompt workup
- Step 1 - History & physical (key clues: aquagenic pruritus, splenomegaly, smoking, drugs)
- Step 2 - Repeat CBC after hydration to rule out relative polycythemia / Gaisböck syndrome
- Step 3 - ABG + co-oximetry (not just pulse ox) to check for hypoxic cause
- Step 4 (Pivotal) - Serum EPO level branches the entire workup:
- Low EPO → PV pathway (JAK2, bone marrow biopsy, WHO criteria)
- High EPO → Secondary pathway (smoker's polycythemia via COHb, paraneoplastic tumours, genetic/high-affinity Hb causes)
- Red alert box - Urgent indications for immediate phlebotomy and complications to watch for
How testosterone, sglt2 inhibitors cause polycythemia
SGLT2 inhibitor erythrocytosis hematocrit mechanism
SGLT2 inhibitor erythropoietin hemoglobin increase mechanism
PMID: 36657305
PMID: 38604895
testosterone polycythemia mechanism EPO erythropoiesis direct indirect
How Testosterone and SGLT2 Inhibitors Cause Polycythemia
Part 1: Testosterone-Induced Polycythemia
Epidemiology
Mechanisms - Multiple Pathways
1. EPO Stimulation (Indirect - via kidneys)
- Testosterone acts on renal peritubular cells to stimulate EPO gene transcription
- This raises the homeostatic EPO "set point" - more EPO is produced at any given oxygen level
- EPO then acts on CFU-E (Colony Forming Unit-Erythroid) progenitors to drive red cell proliferation, differentiation, and survival
- Note: The EPO level in testosterone-induced polycythemia is variable - it may be transiently elevated, then suppressed once Hb rises (normal feedback), making it an unreliable marker
2. Hepcidin Suppression -> Increased Iron Availability
- Testosterone suppresses hepcidin, the master regulator of iron metabolism
- Low hepcidin increases ferroportin activity on enterocytes and macrophages -> more iron released into circulation
- Greater iron bioavailability removes the rate-limiting step for haemoglobin synthesis and erythropoiesis
- This is now considered one of the most important mechanisms
3. Direct Bone Marrow Stimulation
- Testosterone (and its active metabolite dihydrotestosterone/DHT) acts directly on androgen receptors on erythroid precursors in the bone marrow
- Increases the number of EPO-responsive cells (BFU-E and CFU-E progenitors) in the marrow
- This is EPO-independent - erythropoiesis is amplified even without a rise in EPO
4. Aromatization to Estradiol -> Erythroid Stimulation
- In adipose tissue (especially in obese patients), testosterone is aromatized to estradiol
- Estradiol stimulates hematopoietic stem cell proliferation via estrogen receptor alpha (ERα)
- This explains why higher BMI is an independent risk factor for testosterone-induced erythrocytosis
5. IGF-1 Mediation
- Testosterone may increase insulin-like growth factor 1 (IGF-1), which has direct erythropoietic effects
Physiologic Basis
"Erythropoiesis increases [at puberty], resulting in higher hematocrit and hemoglobin concentrations in men than boys or women." - Goodman & Gilman's Pharmacological Basis of Therapeutics
Clinical Management of Testosterone-Induced Erythrocytosis
| Threshold | Action |
|---|---|
| Baseline Hct ≥50% | Contraindication to starting testosterone |
| Hct rises to >54% on TRT | Withhold testosterone, investigate, consider dose reduction or phlebotomy |
| Monitoring | CBC at 3-6 months after starting, then annually |
- Risk is highest with injectable esters (peak-trough fluctuations); lower with transdermal gels; lowest with pellets
Part 2: SGLT2 Inhibitor-Induced Erythrocytosis
The Debate: Hemoconcentration vs. True Erythropoiesis
| Evidence AGAINST simple hemoconcentration | Evidence FOR true erythropoiesis |
|---|---|
| Diuretic-class drugs (furosemide) cause long-term worsening of renal function | SGLT2i preserve renal function long-term |
| Hemoconcentration would raise albumin, BUN proportionally | These markers do not rise proportionately |
| Plasma volume contraction is transient; Hb rise with SGLT2i is sustained | Hb rise persists at 6-12 months |
| The Hb increase is the best predictor of cardiorenal benefit - a diuretic effect alone would not explain organ protection | EPO levels measurably increase with SGLT2i use |
The True Mechanism: Amelioration of Renal Hypoxia
-
The diabetic/CKD kidney is chronically hypoxic
- High glucose load forces the proximal tubule to work overtime reabsorbing glucose via SGLT2, consuming excessive oxygen
- The proximal tubule sits in a borderline hypoxic zone of the renal cortex even normally (low O₂ tension, high metabolic demand)
- In diabetes + CKD, this hypoxia is worsened
-
SGLT2 inhibition reduces proximal tubular workload
- By blocking SGLT2, glucose reabsorption in the proximal tubule is reduced -> less oxygen consumed per nephron
- Renal cortex hypoxia is ameliorated
-
Improved renal oxygenation -> more efficient EPO production
- EPO is produced by peritubular fibroblasts in the renal cortex, triggered by HIF-1α/HIF-2α when O₂ tension falls
- Paradoxically, restoring normal O₂ sensing (by reducing the pathological hypoxia suppressing EPO in diseased kidneys) allows EPO to rise back toward normal
- This increased EPO drives genuine erythropoiesis - a true increase in red cell mass
-
Additional: Iron metabolism improvement
- SGLT2i may reduce inflammation (lowering hepcidin indirectly) -> improved iron bioavailability for erythropoiesis
Why This Matters Clinically
"The increase in hematocrit with SGLT2i treatment is not due to hemoconcentration, but to an increase in erythropoiesis due to amelioration of hypoxia and more efficient erythropoietin production." - Ekanayake & Mudaliar, Diabetes Metab Syndr, 2023
Is This "Polycythemia" Clinically Concerning?
Summary Comparison
| Feature | Testosterone | SGLT2 Inhibitors |
|---|---|---|
| Type | True absolute (increased red cell mass) | True absolute (increased erythropoiesis) + initial mild relative |
| EPO role | Stimulated + hepcidin-mediated iron availability + direct marrow effect | Restored via amelioration of renal hypoxia |
| Hepcidin | Suppressed (key mechanism) | Indirectly reduced via anti-inflammatory effects |
| Magnitude | Can be severe (Hct >60%); dose-dependent | Modest (~0.5-1 g/dL Hb); rarely clinically problematic |
| Clinical concern | VTE risk, thrombosis; requires monitoring and dose adjustment | Generally beneficial; cardio-renal protective mechanism |
| Management | Monitor Hct; hold/reduce dose if Hct >54%; phlebotomy if needed | No specific intervention needed; continue drug |
- Harrison's Principles of Internal Medicine 22e
- Goodman & Gilman's Pharmacological Basis of Therapeutics
- Ekanayake & Mudaliar, Diabetes Metab Syndr 2023, PMID 36657305
- Cases et al., Nefrologia 2024, PMID 38604895
Why raised LDH and uric acid
Why LDH and Uric Acid Are Raised in Polycythemia Vera
Raised LDH - The Mechanism
What is LDH?
Why Does It Rise in PV?
- Many of these cells undergo ineffective haematopoiesis - they proliferate but are destroyed in the marrow before reaching the circulation (intramedullary cell death)
- Mature cells have a normal lifespan, but with greatly expanded numbers, the absolute daily cell death is far higher
- Splenic sequestration and destruction is increased due to splenomegaly
- Vascular thromboses cause local cell necrosis
LDH as a Marker in MPNs
- Elevated LDH at diagnosis correlates with higher disease burden and more aggressive disease
- A rising LDH in PV can signal progression to spent-phase myelofibrosis or blast crisis (AML transformation) - both involve massive cell turnover/necrosis
The general principle: LDH rises in any condition of rapid cell turnover or cell death - haemolysis, leukaemia, lymphoma, solid tumours, and tissue infarction. In PV it is the neoplastic cell proliferation + turnover that drives it.
Raised Uric Acid (Hyperuricaemia) - The Mechanism
The Purine Pathway
Cell DNA/RNA (adenine, guanine)
|
| [Cell death -> nucleic acid release]
↓
Purines (AMP, GMP)
|
| Hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
↓
Hypoxanthine / Xanthine
|
| Xanthine oxidase
↓
URIC ACID <--- accumulates in plasma
Why Does It Rise in PV?
- Every dividing cell contains purine-rich DNA and RNA (adenine + guanine bases)
- In PV, the JAK2 clone continuously produces and destroys enormous numbers of cells
- As these cells die and their nuclei are degraded, huge quantities of purines are released
- These are catabolised through the xanthine oxidase pathway to uric acid
- The rate of uric acid production overwhelms renal excretory capacity -> secondary hyperuricaemia
- Erythroid cells (most numerous) - large nuclear DNA during precursor stages
- Granulocytes - rich in nuclear DNA
- Megakaryocytes - polyploid (massively enlarged nuclei with multiple genome copies), releasing huge purine loads when they fragment
| Uric acid level | Risk of gout per year |
|---|---|
| 7.0-8.9 mg/dL | ~0.5% |
| >9.0 mg/dL | ~5% |
- Textbook of Family Medicine 9e, Table 14-18: "Polycythemia vera" explicitly listed under Overproduction causes of hyperuricaemia (alongside myeloproliferative disorders, malignancies, haemolytic anaemia)
Summary: Common Root Cause
| Lab Finding | Molecule | Source | Root Mechanism |
|---|---|---|---|
| Elevated LDH | Lactate dehydrogenase | Released from lysed/dying cells | Massively expanded neoplastic cell mass + high turnover (ineffective haematopoiesis, senescence, infarction) |
| Elevated Uric Acid | End-product of purine catabolism (via xanthine oxidase) | Degraded DNA/RNA from dying nuclei | Same high cell turnover -> massive purine nucleotide release -> xanthine oxidase -> uric acid overproduction |
Why platelets dysfunction
platelet dysfunction polycythemia vera mechanisms receptor defects acquired von Willebrand thrombosis bleeding paradox
Why Platelets Are Dysfunctional in Polycythemia Vera
The Core Problem
Mechanism 1: Intrinsic Receptor Defects (Clonal Origin)
| Receptor | Normal Function | Defect in PV |
|---|---|---|
| Thromboxane A2 receptor (TP) | Amplifies platelet activation, promotes aggregation | Reduced expression / impaired signalling |
| ADP receptor (P2Y12) | Key amplification signal for aggregation | Reduced density or impaired response |
| PAR-1 (thrombin receptor) | Thrombin-mediated activation | Impaired in some clonal platelets |
| Collagen receptor (GPVI) | Triggers activation at site of vessel injury | Abnormal in neoplastic clone |
Mechanism 2: Arachidonic Acid / Thromboxane Pathway Defect
- The AA -> TxA2 signalling pathway is constitutively hyperactivated at baseline (due to JAK2-driven signalling), leading to chronic low-grade platelet activation
- This chronic pre-activation depletes platelets of their activation machinery - they become "exhausted" and cannot respond to a fresh stimulus properly
- Giant platelets and megakaryocyte fragments seen in PV blood have abnormal cytoskeletal organisation, further impairing shape change and release reactions
Mechanism 3: Granule Abnormalities
- Dense granules (δ-granules): contain ADP, ATP, serotonin, Ca²⁺ - amplify aggregation
- Alpha granules (α-granules): contain fibrinogen, VWF, P-selectin, coagulation factors - promote adhesion and clot formation
- Dense granule content is reduced or depleted (partial dense granule deficiency)
- Alpha granule release is impaired
- This means the normal positive-feedback amplification of platelet plug formation is blunted -> bleeding tendency
Mechanism 4: Acquired von Willebrand Syndrome (AvWS) - The Key to the Bleeding Paradox
Normal VWF Function
How Extreme Thrombocytosis Destroys HMW VWF Multimers
Massive thrombocytosis in PV/ET
|
↓
Enormous total platelet surface area
(GPIb receptors on each platelet)
|
↓
In vivo adsorption of HMW VWF multimers
onto platelet surface GPIb receptors
|
↓
HMW multimers are rapidly cleared
from circulation (along with platelets)
|
↓
Selective loss of HMW VWF multimers
(similar pattern to Type 2A VWD)
|
↓
Impaired platelet adhesion at high shear
(mucocutaneous bleeding: epistaxis,
GI bleeding, bruising, menorrhagia)
This is why PV can cause both thrombosis (from activated clonal platelets + hyperviscosity) AND bleeding (from AvWS + platelet dysfunction) - sometimes in the same patient.
Mechanism 5: JAK2-Driven Paradoxical Hyperactivation -> Thrombosis
- JAK2 V617F in platelets causes constitutive JAK-STAT signalling even without external stimulation
- This produces a basally hyperactivated state: increased P-selectin expression, increased platelet-leukocyte aggregates, increased tissue factor expression
- The viscous, hypercellular blood has slow flow -> amplifies thrombotic risk further
- Result: thrombosis in unusual sites - hepatic vein (Budd-Chiari syndrome), portal vein, mesenteric veins, cerebral sinus
The Paradox Explained: Same Platelets, Opposite Outcomes
| Condition | Mechanism | Clinical Result |
|---|---|---|
| Platelet count moderately elevated | JAK2-hyperactivated platelets, intact VWF | Thrombosis dominates |
| Platelet count extremely high (>1,000-1,500 × 10⁹/L) | AvWS - HMW VWF multimers consumed | Bleeding dominates |
| Giant platelets/megakaryocyte fragments | Structural and granule defects | Impaired primary haemostasis -> bleeding |
"Platelets produced from the neoplastic clone are often dysfunctional, a derangement that contributes to the elevated risk of thrombosis and bleeding." - Robbins & Kumar Basic Pathology
- Robbins & Kumar Basic Pathology (Robbins Pathology)
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Michiels et al., Semin Thromb Hemost 2006 - The paradox of platelet activation and impaired function in ET and PV
- AvWS in myeloproliferative neoplasms, Cancers (Basel) 2020, PMC7407619