Embolism pathology
Reading File
Finding Sources
Finding Sources
Searching PubMed
"embolism"[MeSH Terms] AND "pathology"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Excellent content retrieved. I now have comprehensive material on all types of embolism. Let me compile the complete answer.
Embolism - Pathology
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease; Robbins & Kumar Basic Pathology
Definition
An embolus is a detached intravascular solid, liquid, or gaseous mass that is carried by the blood from its point of origin to a distant site, where it causes tissue dysfunction or infarction. The vast majority of emboli derive from dislodged thrombi - hence the term thromboembolism. Less commonly, emboli consist of fat droplets, air/nitrogen bubbles, atherosclerotic debris (cholesterol emboli), tumor fragments, bone marrow bits, or amniotic fluid. Emboli lodge in vessels too small for further passage, causing partial or complete vascular occlusion.
Types of Embolism
1. Pulmonary Thromboembolism (PE)
Source: >95% originate from deep venous thrombi (DVT) of the legs - specifically proximal to the popliteal fossa. Incidence: 60-120 cases/100,000 per year in the USA; causes 60,000-100,000 deaths/year.
Morphology and Travel:
- Fragmented DVT thrombi travel through progressively larger veins, pass through the right heart, and arrest in the pulmonary vasculature
- A large embolus can occlude the main pulmonary artery
- A saddle embolus straddles the pulmonary artery bifurcation (Fig. 4.15 below)
- Smaller emboli pass into branching arterioles
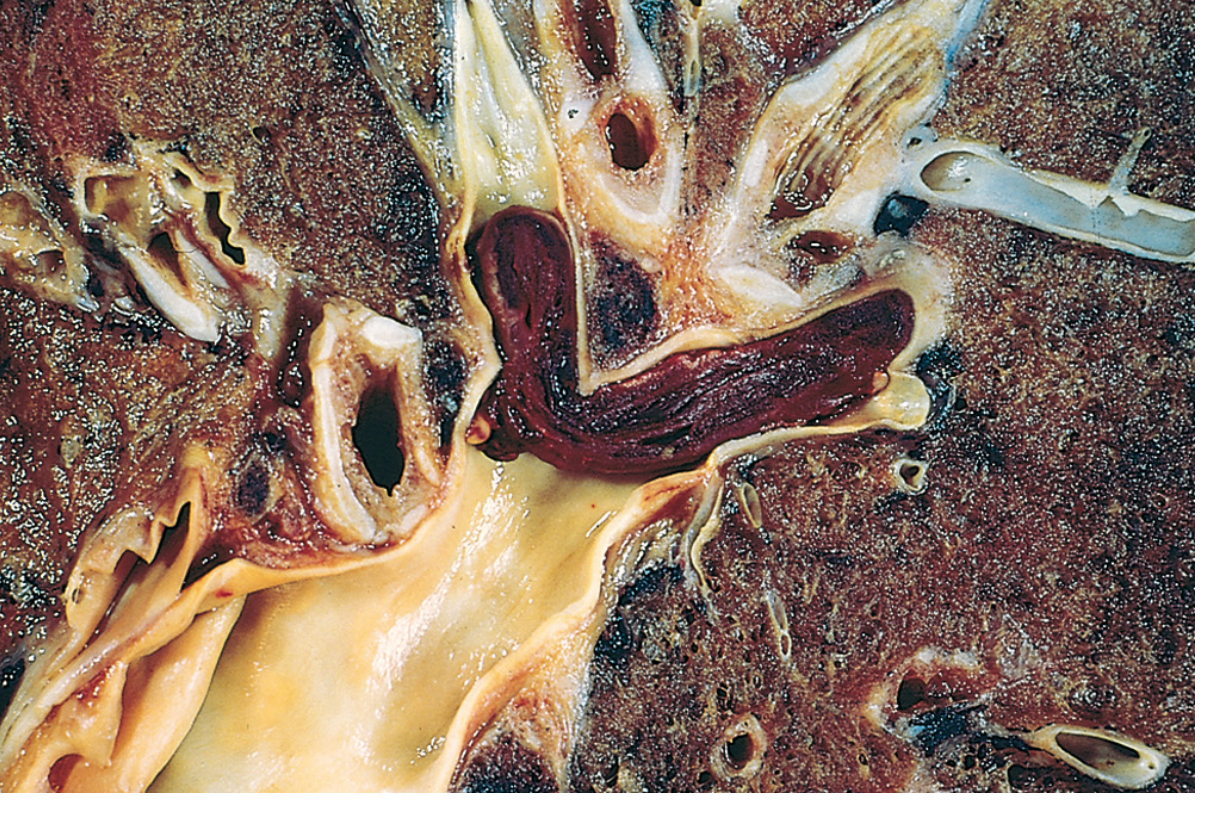
Functional Consequences (by size):
| Embolus Size | Consequence |
|---|---|
| Small (60-80% of cases) | Clinically silent; undergo organization, incorporated into vessel wall; may leave bridging fibrous webs |
| Medium | Pulmonary hemorrhage (usually NOT infarction - dual bronchial/pulmonary circulation protects); infarction occurs if bronchial flow is compromised (e.g., left heart failure) |
| Small end-arteriolar | Often causes infarction or hemorrhage |
| Large (>60% pulmonary circulation blocked) | Sudden death, acute right-heart failure (cor pulmonale), cardiovascular collapse |
| Recurrent multiple | Pulmonary hypertension + right ventricular failure (cor pulmonale) |
Paradoxical embolism: Venous embolus passes through an atrial or ventricular septal defect into the systemic arterial circulation.
2. Systemic Thromboembolism
Sources:
- 80% arise from intracardiac mural thrombi
- Two-thirds: left ventricular wall infarcts
- One-fourth: left atrial dilation and fibrillation
- Remainder: aortic aneurysms, atherosclerotic plaques, valvular vegetations, venous thrombi (paradoxical)
- 10-15%: unknown origin
Distribution of arrest sites:
- Lower extremities: 75%
- Brain: 10%
- Others: intestines, kidneys, spleen, upper extremities
Consequences: Tissue infarction; depends on vulnerability of tissue to ischemia, caliber of occluded vessel, and presence/absence of collateral circulation.
3. Fat Embolism
Cause: Microscopic fat globules (sometimes with hematopoietic bone marrow) enter the vasculature after:
- Fractures of long bones (most common)
- Rarely: soft tissue trauma and burns
Occurs in ~90% of individuals with severe skeletal injuries; vascular sinusoids/venules in marrow rupture, allowing marrow and adipose tissue to herniate into vessels.
Fat Embolism Syndrome (symptomatic minority):
- Onset: 1-3 days after injury
- Classic triad: Pulmonary insufficiency + Neurologic symptoms + Anemia/thrombocytopenia
- Acute features: tachypnea, dyspnea, tachycardia, irritability, restlessness, delirium/coma
- Petechial rash (20-50% of cases) - diagnostic clue, due to rapid thrombocytopenia
- Fatal in 5-15% of cases
Pathogenesis - two mechanisms:
- Mechanical obstruction: Fat microemboli + RBC/platelet aggregates occlude pulmonary and cerebral microvasculature
- Biochemical injury: Free fatty acids released from fat globules cause toxic endothelial injury; platelet activation and granulocyte recruitment (free radicals, proteases, eicosanoids)
Histology note: Fat is dissolved by solvents used in paraffin embedding; fat microglobules must be demonstrated with frozen sections (Oil Red O stain).
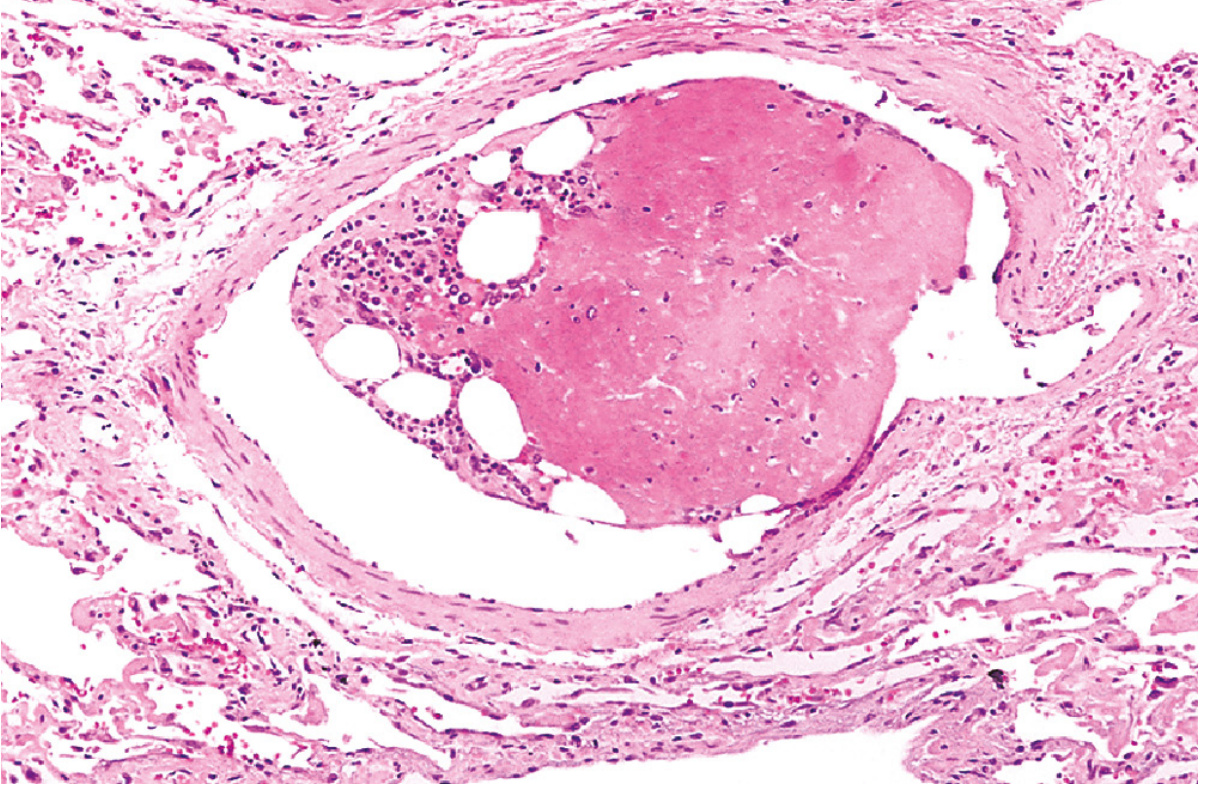
4. Air Embolism (Gas Embolism / Decompression Sickness)
Mechanism: Rapid decompression (most commonly in scuba divers ascending too quickly) causes sudden formation of nitrogen gas bubbles within the vasculature and tissues.
Pathogenesis: Gas bubbles form in skeletal muscles and joint spaces (causing the "bends" - agonizing pain) and can embolize to the pulmonary vasculature, brain, and coronary circulation.
Clinical effects:
- Focal ischemia in affected organs
- Pulmonary edema ("chokes")
- Stroke-like manifestations
- In caisson workers and divers: chronic form leads to multifocal ischemic necrosis of bone (femoral heads, tibia, humeri)
Treatment: Hyperbaric oxygen chamber (recompression forces gases back into solution).
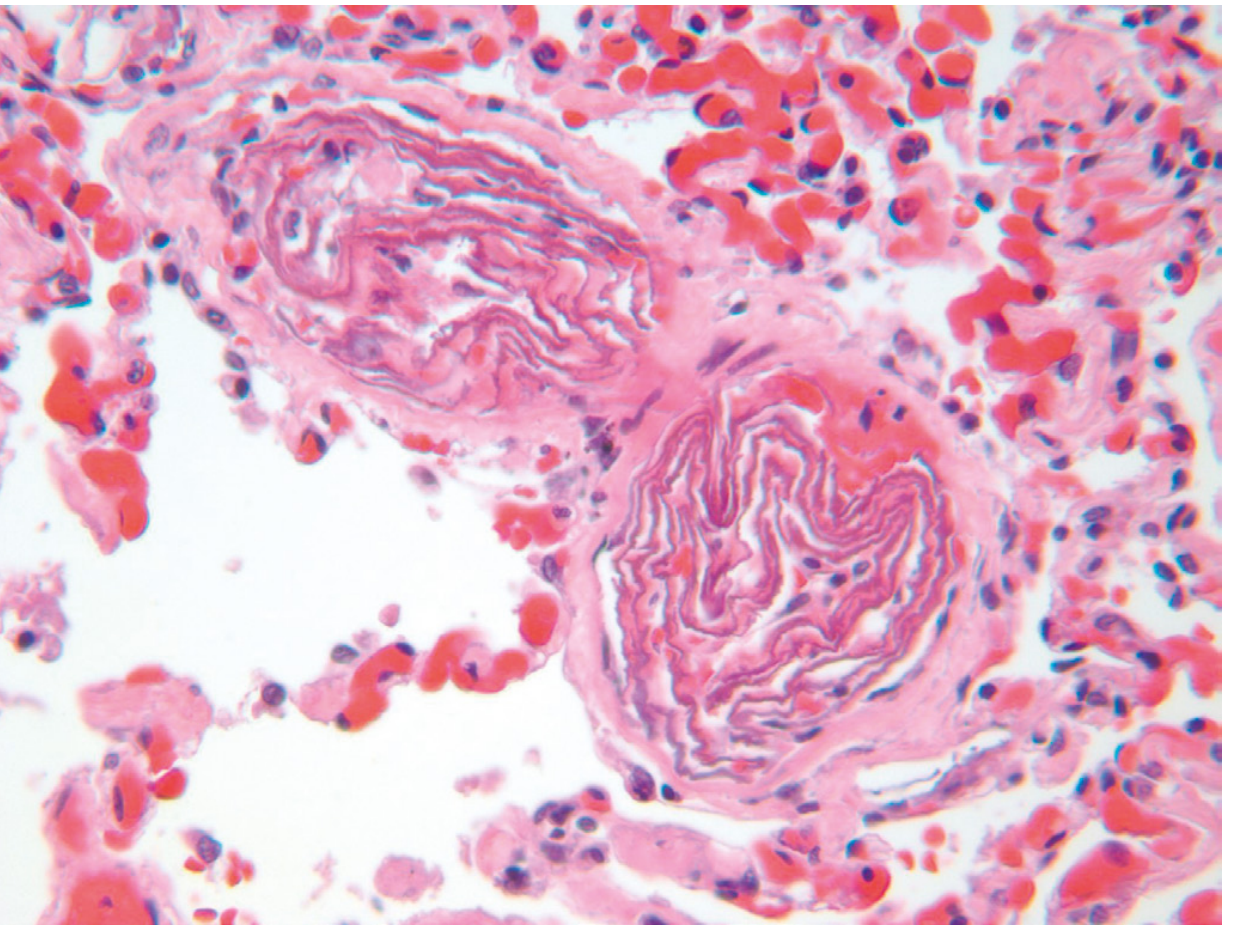
5. Amniotic Fluid Embolism
Cause: Rare but often fatal complication of childbirth/peripartum period; amniotic fluid (with fetal cells and debris) enters the maternal circulation through tears in placental membranes or uterine veins.
Pathogenesis: Squamous cells, lanugo hair, fat from vernix, and other fetal contents enter maternal pulmonary vasculature, triggering:
- Severe pulmonary dysfunction
- DIC (from thrombogenic material in amniotic fluid)
- Cardiovascular collapse
Histology (pathognomonic): Small pulmonary arterioles packed with laminated swirls of fetal squamous cells + pulmonary edema + congestion + small organizing thrombi (DIC)
Key Concepts Summary
| Type | Source | Key Target | Hallmark Feature |
|---|---|---|---|
| Pulmonary thromboembolism | DVT (leg veins) | Pulmonary arteries | Saddle embolus; 60-80% silent |
| Systemic thromboembolism | Cardiac mural thrombi (LV/LA) | Limbs, brain | Arterial infarction |
| Fat embolism | Long bone fractures | Lung, brain | Petechiae; fat globules on frozen section |
| Air embolism | Decompression | Joints, lung, brain | "Bends"; nitrogen bubbles |
| Amniotic fluid embolism | Parturition | Pulmonary arterioles | Fetal squamous cells; DIC |
Recent literature note: A 2025 systematic review (PMID 39478415) on fat embolism pathology emphasizes the need for standardized histopathological procedures for its diagnosis, particularly regarding appropriate tissue fixation and staining methods - consistent with the textbook guidance on frozen sections. A 2025 systematic review on cadaver clots (PMID 40029560) addresses post-mortem distinction of thromboemboli from cadaveric clots, relevant in forensic pathology.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 130-133
- Robbins & Kumar Basic Pathology, pp. 93-95
Apoptosis pathology
Finding Sources
Finding Sources
Reading File
Reading File
Searching PubMed
"apoptosis"[MeSH Terms] AND "pathology"[MeSH Terms]
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Finding Sources
Loading Image
Reading File
I now have everything needed for a complete, comprehensive answer on apoptosis pathology. Let me compile it.
Apoptosis - Pathology
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease; Robbins & Kumar Basic Pathology
Definition
Apoptosis is a regulated, programmed form of cell death in which cells activate intrinsic enzymes (caspases) that degrade cellular DNA and nuclear and cytoplasmic proteins. The dying cell fragments into plasma membrane-bound fragments called apoptotic bodies, which are rapidly phagocytosed before their contents leak out - meaning apoptosis does not elicit an inflammatory reaction, in stark contrast to necrosis.
The word "apoptosis" comes from the Greek for "falling off" (like leaves from a tree), first described morphologically in 1972. It was subsequently discovered in model organisms (e.g., C. elegans) where certain cells die at precise times during development - the phenomenon called programmed cell death.
Apoptosis vs. Necrosis
| Feature | Apoptosis | Necrosis |
|---|---|---|
| Mechanism | Regulated, caspase-driven | Uncontrolled |
| Cell size | Shrinkage | Swelling (oncosis) |
| Plasma membrane | Intact | Disrupted |
| DNA degradation | Internucleosomal laddering (regular fragments) | Random degradation |
| Inflammation | None (contents contained) | Yes (contents leak out) |
| Energy requirement | Active (ATP required) | Passive |
| Outcome | Phagocytosis of apoptotic bodies | Inflammatory lysis |
Causes of Apoptosis
Physiologic (normal) situations
Apoptosis is a critical homeostatic mechanism - humans turn over approximately 1 million cells per second. It occurs in:
- Embryogenesis - removal of supernumerary cells, involution of primordial structures, remodeling of maturing tissues (programmed cell death)
- Hormone-dependent involution - endometrial breakdown in the menstrual cycle, ovarian follicular atresia at menopause, regression of the lactating breast after weaning
- Cell turnover - e.g., intestinal crypt epithelium, to maintain constant cell numbers (homeostasis)
- Lymphocyte elimination:
- Immature lymphocytes in bone marrow and thymus that fail to produce functional antigen receptors
- Germinal center B cells that fail to produce high-affinity antibodies
- Self-reactive lymphocytes (prevents autoimmunity)
- Spent effector cells - neutrophils after acute inflammation, lymphocytes at the end of an immune response
Pathologic situations
Apoptosis eliminates irreparably damaged cells without causing collateral damage:
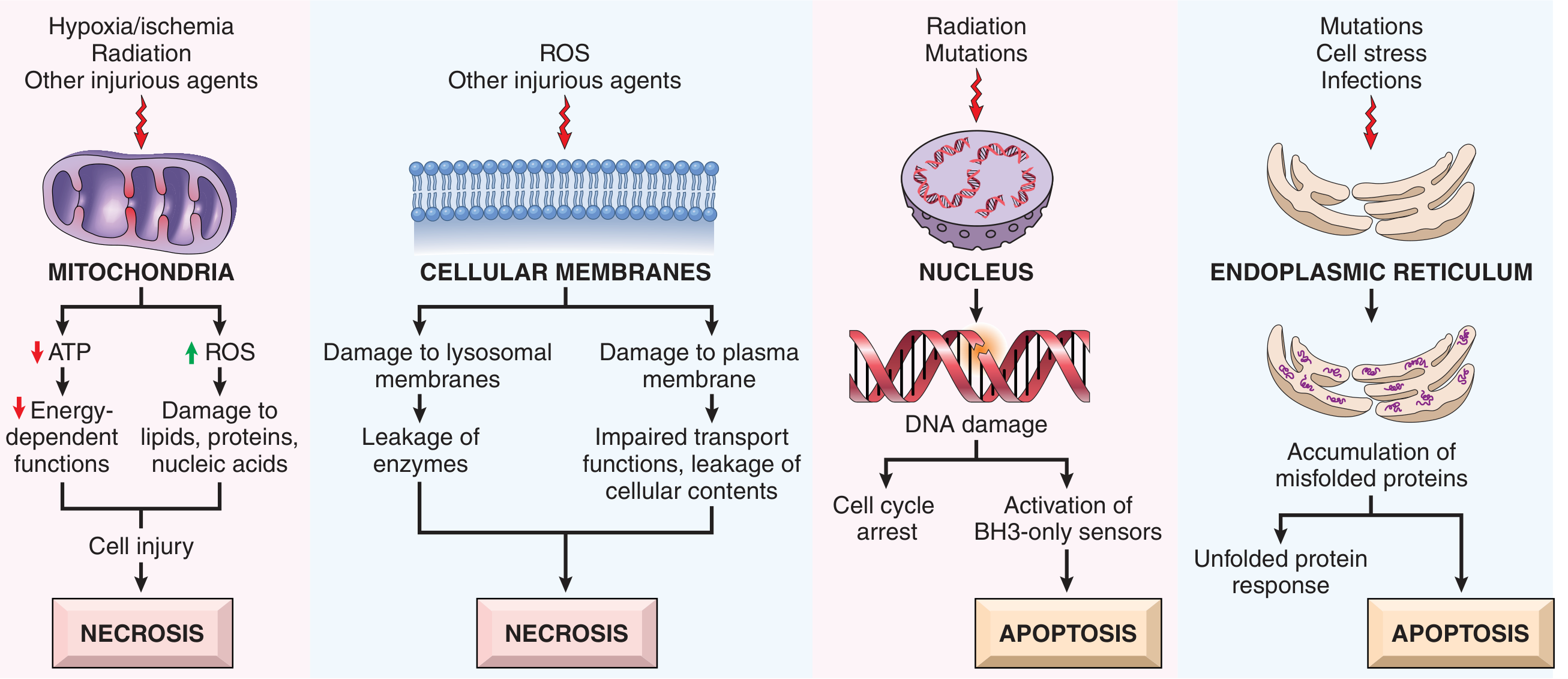
- DNA damage - radiation, cytotoxic drugs (directly or via free radicals) → activates BH3-only proapoptotic proteins → if irreparable, triggers apoptosis
- Accumulation of misfolded proteins (ER stress) - activates BH3-only proteins ± direct caspase activation
- Viral infections - viral proteins activate proapoptotic molecules; infected cells killed by cytotoxic T lymphocytes (CTLs) via the death receptor pathway
- Pathologic atrophy - e.g., parenchymal organ atrophy after duct obstruction
Morphologic and Biochemical Changes
Morphology
- Cell shrinkage - cell is smaller, cytoplasm dense, organelles compacted
- Chromatin condensation (pyknosis) - dense crescents of chromatin peripherally aggregated against the nuclear membrane
- Nuclear fragmentation (karyorrhexis) - condensed chromatin breaks up
- Cytoplasmic blebs and apoptotic bodies - membrane blebbing produces fragments containing portions of cytoplasm ± nuclear material; membrane remains intact
- Phagocytosis - apoptotic bodies display "find me" signals (e.g., fractalkine, ATP) and "eat me" signals (e.g., phosphatidylserine flipped to the outer leaflet) recognized by macrophages → rapid engulfment
- No inflammation - contents never released
Biochemistry
- Caspase activation - proteases with cysteine in the active site, cleave after aspartate residues; exist as inactive proenzymes
- DNA laddering - internucleosomal cleavage generates DNA fragments of ~180 bp multiples (ladder pattern on gel electrophoresis)
- Phosphatidylserine externalization - detected by Annexin V staining (used to identify apoptotic cells)
Mechanisms of Apoptosis - Two Pathways
Both pathways converge on caspase activation, divided into:
- Initiation phase - initiator caspases become active
- Execution phase - executioner caspases (e.g., caspase-3) drive cell fragmentation
Pathway 1: Mitochondrial (Intrinsic) Pathway
The predominant pathway in most physiologic and pathologic apoptosis.
Trigger: Loss of survival signals, DNA damage, or ER stress (misfolded proteins)
BCL2 Family - the master regulators:
| Group | Members | Domains | Function |
|---|---|---|---|
| Anti-apoptotic | BCL2, BCL-XL, MCL1 | BH1-4 | Keep outer mitochondrial membrane impermeable; prevent cytochrome c release |
| Pro-apoptotic effectors | BAX, BAK | BH1-3 | Oligomerize in outer mitochondrial membrane → pores → cytochrome c leaks out |
| BH3-only sensors | BAD, BIM, BID, PUMA, NOXA | BH3 only | Sensors of stress; when activated, directly activate BAX/BAK OR neutralize BCL2/BCL-XL |
Sequence of events:
- Stress/damage → BH3-only proteins upregulated (increased transcription + posttranslational modification)
- BH3-only proteins activate BAX/BAK → oligomerize in outer mitochondrial membrane
- Simultaneously, survival signals fall → BCL2/BCL-XL synthesis declines
- Cytochrome c (and Smac/DIABLO) released from mitochondrial intermembrane space into cytoplasm
- Cytochrome c + APAF-1 (apoptosis-activating factor-1) form the apoptosome (multimeric "wheel" complex)
- Apoptosome recruits and activates caspase-9 (initiator caspase) by autocatalytic cleavage
- Caspase-9 cleaves and activates caspase-3 (executioner) → cell death
- Smac/DIABLO neutralizes IAPs (inhibitors of apoptosis proteins), removing the brake on caspase activation
BCL2 overexpression (due to t(14;18) chromosomal translocation in follicular lymphoma) is the prototypic example of cancer survival through apoptosis evasion.
Pathway 2: Death Receptor (Extrinsic) Pathway
Primarily used to eliminate self-reactive lymphocytes and by CTLs to kill infected/tumor cells.
Key death receptors: Members of the TNF receptor superfamily with a cytoplasmic death domain
- Fas (CD95) - the best-characterized
- TNFR1 (type 1 TNF receptor)
Sequence of events (using Fas as example):
- FasL (expressed on self-reactive T cells and some CTLs) binds Fas on target cells
- Three or more Fas molecules cluster → their death domains bind adaptor protein FADD (Fas-associated death domain)
- FADD recruits inactive caspase-8 (or -10) → multiple caspase molecules brought together → autocatalytic cleavage → active caspase-8
- Active caspase-8 directly cleaves caspase-3 (executioner) → cell death
- Amplification via BID cleavage: caspase-8 cleaves BID (a BH3-only protein) → truncated BID (tBID) activates BAX/BAK → crossover to the mitochondrial pathway
Execution Phase (Common Final Pathway)
Once executioner caspases (especially caspase-3) are activated:
- Activate CAD (caspase-activated DNase) → internucleosomal DNA cleavage
- Cleave cytoskeletal and nuclear scaffold proteins → cell shrinkage and nuclear fragmentation
- Activate enzymes that disrupt cell membrane → phosphatidylserine externalization (phagocytic signal)
Apoptosis in Disease - Pathologic Relevance
| Too Little Apoptosis (Insufficient cell death) | Too Much Apoptosis (Excessive cell death) |
|---|---|
| Cancer (BCL2 overexpression, p53 loss) | Neurodegenerative diseases (Parkinson, Alzheimer, ALS) |
| Autoimmune disease (failure to eliminate self-reactive lymphocytes) | Ischemia-reperfusion injury |
| Viral persistence (viral BCL2 homologs, caspase inhibitors) | AIDS (CD4+ T cell depletion) |
| Follicular lymphoma (t(14;18)) | Aplastic anemia |
TP53 and Apoptosis
p53 is a key transcription factor that senses DNA damage. When DNA is irreparably damaged:
- p53 transcriptionally upregulates PUMA and NOXA (BH3-only proteins) → activates intrinsic pathway
- Loss of p53 function (mutation in >50% of human cancers) → cells survive despite severe DNA damage → malignant transformation
Key Concepts Summary
-
Apoptosis = regulated caspase-mediated cell death; no inflammation
-
Two pathways: intrinsic (mitochondrial) via BCL2 family/cytochrome c/apoptosome/caspase-9; extrinsic (death receptor) via Fas-FasL/FADD/caspase-8
-
Both converge on caspase-3 (executioner)
-
BCL2 family is the master switch: anti-apoptotic (BCL2, BCL-XL, MCL1) vs. pro-apoptotic effectors (BAX, BAK) vs. BH3-only sensors (BIM, PUMA, NOXA, BAD, BID)
-
Dysregulation underlies cancer (too little) and degenerative disease (too much)
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 56-62
-
Robbins & Kumar Basic Pathology, pp. 23-28
This is a shared conversation. Sign in to Orris to start your own chat.