Discuss the mechanisms of autoimmune diseases. Describe the etiopathogenesis, pathology and lab diagnosis of SLE.
Reading File
Finding Sources
Finding Sources
Searching PubMed
"systemic lupus erythematosus" AND pathogenesis
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I have comprehensive, authoritative content. Now I'll compose the full detailed answer.
Mechanisms of Autoimmune Diseases
I. Overview: Self-Tolerance and Its Failure
Immunologic tolerance is unresponsiveness to an antigen induced by prior exposure to it. Self-tolerance - tolerance to one's own antigens - is the fundamental property that prevents the immune system from attacking the body's own tissues. When this breaks down, autoimmunity results.
Two broad categories of tolerance mechanisms operate:
A. Central Tolerance
Immature lymphocytes that recognize self antigens in the central lymphoid organs (thymus for T cells, bone marrow for B cells) are either:
- Eliminated by apoptosis (clonal deletion)
- In B cells: undergo receptor editing - switching antigen receptors to new, non-self-reactive specificities
B. Peripheral Tolerance
Mature self-reactive lymphocytes that escape central tolerance encounter self antigens in peripheral tissues, and are restrained by:
- Regulatory T cells (Tregs) - suppress autoreactive T and B cells via IL-10, TGF-β, and direct contact
- Co-inhibitory receptors (CTLA-4 and PD-1) - provide inhibitory signals to block lymphocyte activation
- Anergy - functional inactivation when antigen is presented without co-stimulation (signal 2)
- Apoptosis (activation-induced cell death) via Fas-FasL interactions
II. Mechanisms That Cause Autoimmunity
When tolerance fails, several overlapping mechanisms drive autoimmune disease:
1. Inheritance of Susceptibility Genes
- MHC (HLA) genes regulate antigen presentation; specific HLA alleles are strongly associated with particular autoimmune diseases (e.g., HLA-DR3/DR4 in type 1 diabetes, HLA-DQ in SLE)
- Non-MHC genes include those encoding complement components (C1q, C4), Fc receptors, TLRs, and cytokine signaling molecules
- Genome-wide association studies (GWAS) identify loci involved in lymphocyte signaling and interferon responses
2. Failure of B Cell Tolerance
Defective elimination of self-reactive B cells in the bone marrow or defective peripheral tolerance allows autoreactive B cells to persist and be activated.
3. CD4+ Helper T Cell Activation
Self-reactive CD4+ Th cells specific for nuclear or other self antigens escape tolerance and provide cognate help to autoreactive B cells, resulting in high-affinity class-switched autoantibodies.
4. TLR Engagement by Self Nucleic Acids
Toll-like receptors (TLR7, TLR9) normally detect microbial nucleic acids. In autoimmune settings, self DNA and RNA (released from apoptotic cells or NETs) engage these TLRs on B cells and dendritic cells, driving autoantibody production.
5. Type I Interferon Axis
High levels of circulating type I IFNs (IFN-α/β) are seen in SLE and other conditions. Self nucleic acids engage TLRs on plasmacytoid dendritic cells (pDCs), triggering IFN production. IFNs activate DCs and B cells and amplify helper T cell responses - all promoting pathogenic autoantibody production.
6. Molecular Mimicry
Microbial antigens that share structural similarity with self antigens can activate T or B cells that then cross-react with self tissues (e.g., streptococcal M protein and cardiac myosin in rheumatic fever).
7. Epitope Spreading
An immune response against one self antigen causes tissue damage, releasing previously hidden (cryptic) antigens. Lymphocytes recognizing these new epitopes become activated, broadening and perpetuating the autoimmune response.
8. Defective Clearance of Apoptotic Cells
Apoptotic cells expose nuclear material (histones, DNA) on their surfaces. If phagocytic clearance is defective (e.g., due to C1q deficiency), these materials accumulate and drive anti-nuclear antibody production.
9. Inflammation as an Activating Stimulus
Infections cause tissue injury and activate APCs in tissues, providing costimulatory signals that can break peripheral tolerance by activating otherwise anergic self-reactive T cells.
Systemic vs. Organ-Specific Disease
The nature of the immune response determines the pattern:
- Autoantibody-mediated diseases: SLE (immune complex deposition), Graves' disease (anti-TSH receptor stimulating antibodies), myasthenia gravis (anti-AChR)
- T cell-mediated diseases: type 1 diabetes, multiple sclerosis, rheumatoid arthritis
- Mixed: Rheumatoid arthritis involves both anti-CCP antibodies and Th1/Th17 responses
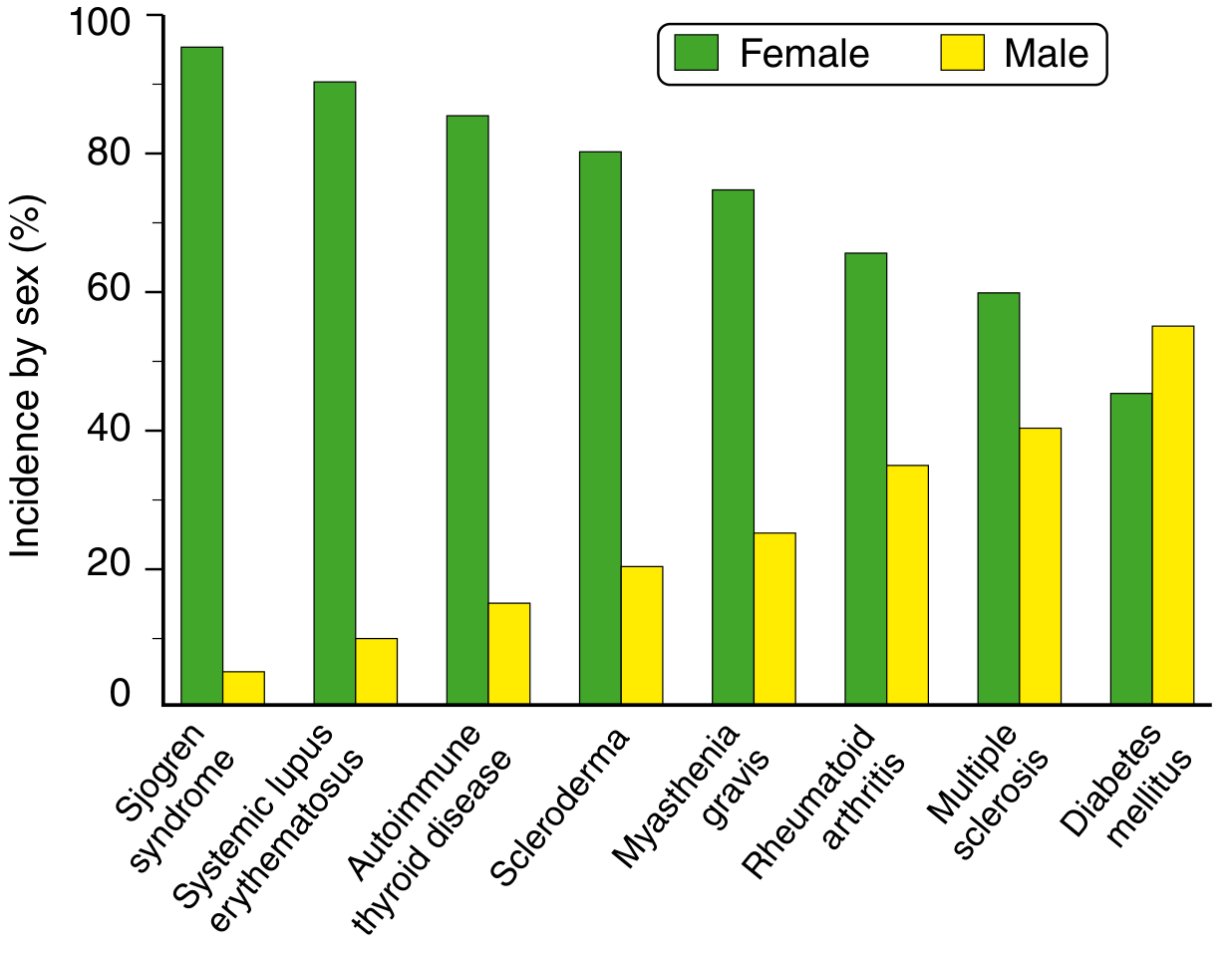
Most systemic autoimmune diseases are chronic and relapsing because self antigens are always present, perpetuating immune activation. They predominantly affect females (see chart below), likely due to sex hormone effects on immune regulation (estrogen enhances B cell responses; the X chromosome encodes many immune-related genes).
Systemic Lupus Erythematosus (SLE)
SLE is the prototypical systemic autoimmune disease - a chronic, remitting-relapsing multiorgan disease characterized by a vast array of autoantibodies (particularly antinuclear antibodies), with tissue injury caused mainly by immune complex deposition and direct antibody binding to cells and tissues.
Epidemiology: Prevalence up to 1 in 2500 in some populations. Female:male ratio of 9:1 during reproductive years (17-55), falling to 2:1 in childhood or after age 65. More prevalent and severe in African, Hispanic, and Asian descent compared to European ancestry.
III. Etiopathogenesis of SLE
A. Genetic Factors
- Family clustering: 20% of first-degree relatives of SLE patients have autoantibodies or immune abnormalities even without clinical disease
- Twin concordance: >20% in monozygotic twins vs. 1-3% in dizygotic twins - confirms genetic contribution but also highlights environmental factors
- HLA associations: Specific HLA-DQ alleles are linked to production of anti-dsDNA, anti-Sm, and antiphospholipid antibodies
- Complement gene deficiencies: Inherited deficiencies of C1q, C2, or C4 strongly predispose to SLE. C1q deficiency impairs phagocytic clearance of apoptotic cells; loss of complement receptor signaling reduces B cell tolerance. Knockout mice lacking C4 develop lupus-like disease.
- GWAS loci: Multiple loci encoding proteins in lymphocyte signaling (PTPN22, BLK, STAT4) and interferon pathways are associated with SLE risk
B. Immunologic Mechanisms
The central defect is failure of self-tolerance leading to production of antinuclear autoantibodies. Several interacting mechanisms operate:
| Mechanism | Details |
|---|---|
| Defective B cell tolerance | Self-reactive B cells escape elimination in bone marrow and peripheral checkpoints |
| Autoreactive CD4+ Th cells | Recognize nucleosomal antigens, provide help for high-affinity anti-dsDNA, anti-histone, anti-Sm antibody production |
| TLR engagement | Nuclear DNA/RNA in immune complexes engage TLR7 and TLR9 on B cells and pDCs, driving further autoantibody production |
| Type I IFN axis | IFN-α/β produced by pDCs activate DCs and B cells; IFN signature correlates with disease severity |
| Defective clearance of apoptotic cells | Released nuclear antigens stimulate anti-nuclear immune responses |
| NETosis | Neutrophil extracellular traps (NETs) release chromatin and nuclear proteins that serve as autoantigens and activate pDCs |
C. Environmental Factors
- UV light: Induces apoptosis in skin keratinocytes, altering DNA in ways that enhance TLR recognition; triggers disease flares
- Sex hormones: Estrogen enhances B cell responses and prolongs B cell survival; the X chromosome encodes many immune genes (TLR7 is X-linked, and gene dosage effects may explain female predominance)
- Drugs: Procainamide, hydralazine, and isoniazid can induce a lupus-like syndrome by altering DNA (drug-induced lupus). This form lacks anti-dsDNA and anti-Sm antibodies, and resolves on drug withdrawal.
- Infections: Certain viruses (notably EBV) may trigger SLE by molecular mimicry or by driving B cell activation
D. Mechanism of Tissue Injury
Tissue injury in SLE occurs through two main mechanisms:
1. Immune complex (Type III hypersensitivity):
- Autoantibodies bind nuclear antigens (liberated from apoptotic cells) circulating in blood
- These immune complexes deposit in glomeruli, skin, joints, and serosal surfaces
- Complement activation and neutrophil/macrophage recruitment cause inflammation and fibrinoid necrosis
- Anti-dsDNA antibody levels and complement C3/C4 consumption correlate with disease activity
2. Direct antibody-mediated injury (Type II hypersensitivity):
- Anti-red cell antibodies → hemolytic anemia
- Anti-platelet antibodies → thrombocytopenia
- Anti-lymphocyte antibodies → lymphopenia
- Antiphospholipid antibodies (anti-β2-glycoprotein I) → thromboembolic disease and pregnancy loss (antiphospholipid syndrome)
IV. Pathology (Morphology) of SLE
The morphologic changes are extremely variable, reflecting multiorgan involvement. The most characteristic lesions result from immune complex deposition.
1. Blood Vessels
- Acute necrotizing vasculitis with fibrinoid necrosis of capillaries, small arteries, and arterioles - can occur in any tissue
- Chronic stage: fibrous thickening with luminal narrowing
2. Kidney (Lupus Nephritis)
- Clinically significant in up to 40% of patients
- Glomerulonephritis from subendothelial and mesangial immune complex deposition
- Characteristic "wire-loop" lesion in active disease - thickened glomerular capillary walls due to massive subendothelial deposits
- WHO/ISN-RPS classification: Class I (minimal mesangial) through Class VI (advanced sclerosing)
- Tubulointerstitial nephritis also occurs
3. Skin
- "Butterfly rash" (malar rash): erythema along the bridge of the nose and both cheeks in ~50% of patients; accentuated by sun exposure
- Discoid rash: erythematous plaques with follicular plugging and scaling
- Histology: vacuolar degeneration of the basal layer of the epidermis, dermal edema, perivascular inflammation, fibrinoid vasculitis
- Immunofluorescence ("lupus band test"): deposition of IgG, IgM, and complement (C3) along the dermoepidermal junction - present in both involved and uninvolved skin
4. Joints
- Non-erosive synovitis involving two or more peripheral joints
- Minimal deformity, contrasting sharply with rheumatoid arthritis
- Tenderness and effusion present
5. Cardiovascular System
- Pericarditis: symptomatic or asymptomatic in up to 50%; may result in effusion or constrictive pericarditis
- Libman-Sacks endocarditis: non-bacterial verrucous endocarditis - small (1-3 mm) warty deposits on either surface of valve leaflets (distinctive), particularly mitral and aortic; more common before widespread steroid use
- Myocarditis: mononuclear cell infiltration causing tachycardia and ECG changes
- Coronary atherosclerosis: accelerated atherosclerosis in long-standing SLE, especially with corticosteroid use
6. Serosal Cavities
- Fibrinous or serofibrinous pleuritis and pericarditis, causing chest pain and effusions
- Chronic adhesions and cavity obliteration in later stages
7. CNS
- No specific morphologic lesion is identified
- Non-inflammatory occlusion of small vessels by intimal proliferation (probably due to endothelial antibody/immune complex damage)
- Clinically manifests as seizures, psychosis, cognitive dysfunction
8. Spleen
- Moderately enlarged (periarteriolar fibrosis)
- "Onion skin" lesion: concentric periarterial fibrosis around splenic arterioles - pathognomonic of SLE
9. Lungs
- Pleuritis most common; lupus pneumonitis less frequent
- Pulmonary hypertension and diffuse alveolar hemorrhage occur
V. Laboratory Diagnosis of SLE
A. Antinuclear Antibodies (ANA) - Screening Test
The indirect immunofluorescence assay (IFA) on Hep-2 cells is the recommended screening test. Patient serum is layered on cultured cells and bound antibodies are detected with fluorochrome-labeled anti-Ig antibody. ANAs are positive in 95-100% of SLE patients (high sensitivity), but are not specific (also seen in other autoimmune diseases and in low titers in normal individuals).
Fluorescence patterns and their significance:
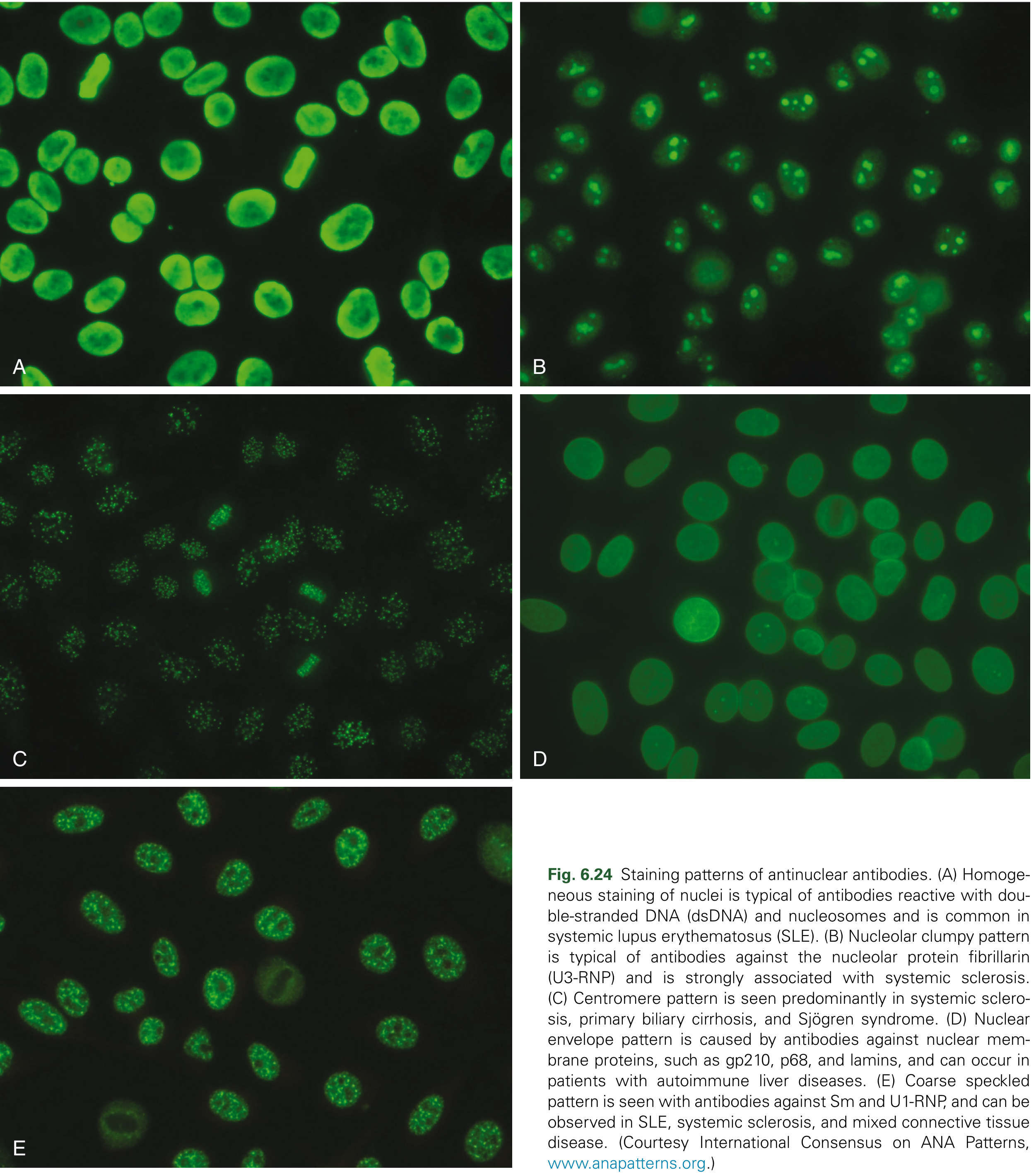
| Pattern | Associated Antibody | Disease Association |
|---|---|---|
| Homogeneous | Anti-dsDNA, anti-histone | SLE (most common) |
| Coarse speckled | Anti-Sm, anti-U1-RNP | SLE, MCTD |
| Fine speckled | Anti-SSA/Ro, anti-SSB/La | SLE, Sjogren's |
| Nucleolar | Anti-Scl-70, anti-fibrillarin | Systemic sclerosis |
| Centromeric | Anti-CENP-A/B/C | Limited scleroderma |
| Rim/peripheral | Anti-gp210, anti-lamin | Primary biliary cholangitis |
| Cytoplasmic dense fine speckled | Anti-ribosomal P | SLE (specific) |
B. Specific Autoantibodies in SLE
| Antibody | Prevalence in SLE | Clinical Significance |
|---|---|---|
| Anti-dsDNA | 40-60% | Specific for SLE; correlates with nephritis activity and complement consumption; used to monitor disease |
| Anti-Sm (Smith) | 20-30% | Highly specific for SLE (diagnostic); target is core proteins of small nuclear RNP particles |
| Anti-histone | Variable | Drug-induced lupus (>95%); also seen in idiopathic SLE |
| Anti-SSA/Ro | Variable | Congenital heart block in neonates; neonatal lupus; Sjögren overlap |
| Anti-SSB/La | Variable | Usually alongside anti-Ro |
| Anti-U1-RNP | 30-40% | Not specific; seen in mixed connective tissue disease |
| Antiphospholipid (anti-β2-GPI) | 30-40% | Antiphospholipid syndrome: venous/arterial thrombosis, recurrent pregnancy loss, thrombocytopenia |
| Anti-ribosomal P | ~10% | Cytoplasmic dense fine speckled pattern; associated with neuropsychiatric lupus |
Key rule: Anti-dsDNA and anti-Sm together are virtually diagnostic of SLE.
C. Complement Studies
- Low C3 and C4 levels during active disease (complement consumed by immune complexes)
- CH50 (total hemolytic complement) also reduced
- Rising complement levels suggest remission; falling levels suggest impending flare
D. Antiphospholipid Antibody Testing
- Lupus anticoagulant (paradoxically causes thrombosis in vivo but prolongs APTT in vitro)
- Anti-cardiolipin IgG/IgM
- Anti-β2-glycoprotein I antibodies
- False-positive VDRL/RPR for syphilis (a classic finding due to antibodies against cardiolipin-lecithin antigen)
E. Routine Laboratory Tests
- CBC: Hemolytic anemia (Coombs-positive), leukopenia (<4,000/mm³ on two or more occasions), lymphopenia (<1,500/mm³), thrombocytopenia
- Urinalysis: Proteinuria (>0.5 g/24h), hematuria, red cell casts (indicate active nephritis)
- ESR and CRP: ESR elevated; CRP often paradoxically normal or only mildly elevated in SLE (distinguishes from infection, where CRP rises sharply)
- Serum creatinine/eGFR: Elevated in renal involvement
- Direct Coombs test: Positive in hemolytic anemia
F. Skin Biopsy - "Lupus Band Test"
Immunofluorescence of skin biopsy shows granular deposition of IgG, IgM, IgA, and C3 along the dermoepidermal junction - positive in both lesional and non-lesional (sun-protected) skin in SLE. Present in non-lesional skin in ~80% of SLE patients (useful for diagnosis).
G. Renal Biopsy
- Indicated when significant proteinuria, active urinary sediment, or rising creatinine is present
- Light microscopy: "wire-loop" lesions, proliferative or membranous patterns
- Immunofluorescence: "Full house" pattern - simultaneous deposits of IgG, IgM, IgA, C3, C1q (characteristic of lupus nephritis)
- Electron microscopy: subendothelial, subepithelial, and mesangial electron-dense deposits
VI. Diagnostic Criteria
The 2019 EULAR/ACR classification criteria (updated from the 1997 ACR criteria) use a weighted scoring system across 10 domains including:
- Constitutional (fever)
- Hematologic (hemolytic anemia, leukopenia, thrombocytopenia)
- Neuropsychiatric (seizures, psychosis)
- Mucocutaneous (malar rash, discoid rash, photosensitivity, oral ulcers)
- Serosal (pleuritis, pericarditis)
- Musculoskeletal (synovitis)
- Renal (proteinuria, RBC casts)
- Antiphospholipid antibodies, complement proteins, SLE-specific antibodies
Entry criterion: ANA positive (≥1:80 titer on Hep-2 cells) is required.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 213-220 | Cellular and Molecular Immunology, 9th ed., p. 956
This is a shared conversation. Sign in to Orris to start your own chat.