Myocardial infarction : gross & microscopy examination, pathogenesis, Etiology, risk factors, diagnosis, complications (early & late), treatment, reperfusion & reperfusion injuriy, intervention, dressler syndrome
Myocardial Infarction - Comprehensive Review
Definition
Etiology & Risk Factors
- Hyperlipidemia (elevated LDL, low HDL)
- Hypertension
- Cigarette smoking
- Diabetes mellitus
- Obesity / sedentary lifestyle
- Stress / type A personality
- Oral contraceptives (increased thrombotic risk)
- Hyperhomocysteinemia
- Advancing age (frequency rises progressively)
- Male sex (females protected during reproductive years; postmenopausal estrogen decline worsens risk)
- Family history of premature CAD
- Prior MI
- Coronary vasospasm (cocaine, ephedrine, Prinzmetal)
- Embolism (mural thrombus in AF, endocarditis vegetations, prosthetic material, patent foramen ovale)
- Vasculitis of coronary arteries
- Hematologic disorders (sickle cell disease, polycythemia)
- Amyloid deposition in vascular walls
- Microembolization / multifocal microinfarction
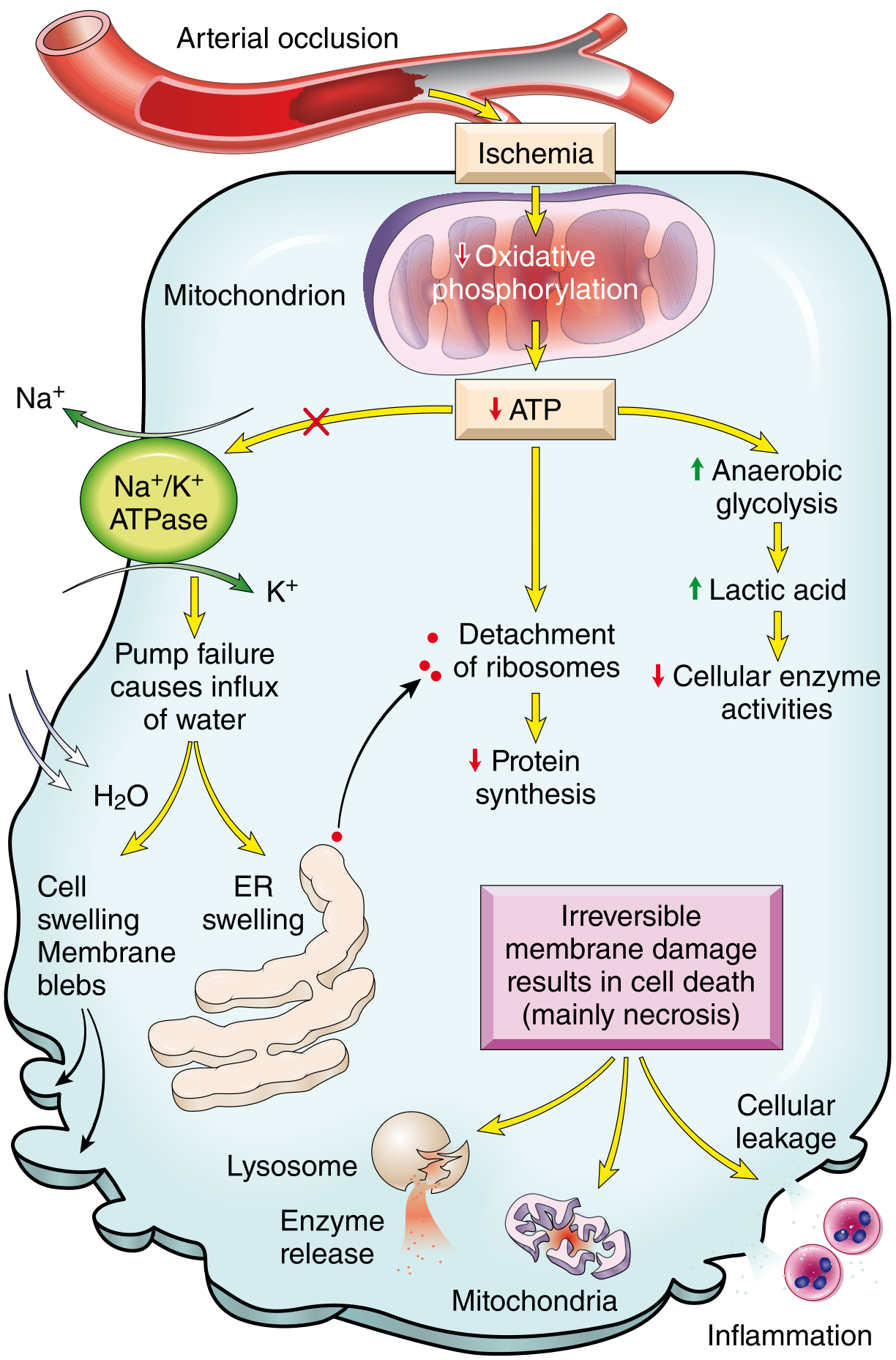
Pathogenesis
| Feature | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | <2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | >1 hour |
- Only severe ischemia (blood flow ≤10% of normal) lasting 20-30+ minutes leads to irreversible necrosis.
- Progressive loss of viability becomes complete by 6-12 hours.
- Necrosis begins in the subendocardial zone (last to receive blood, highest intramural pressure) and progresses outward as a "wavefront."
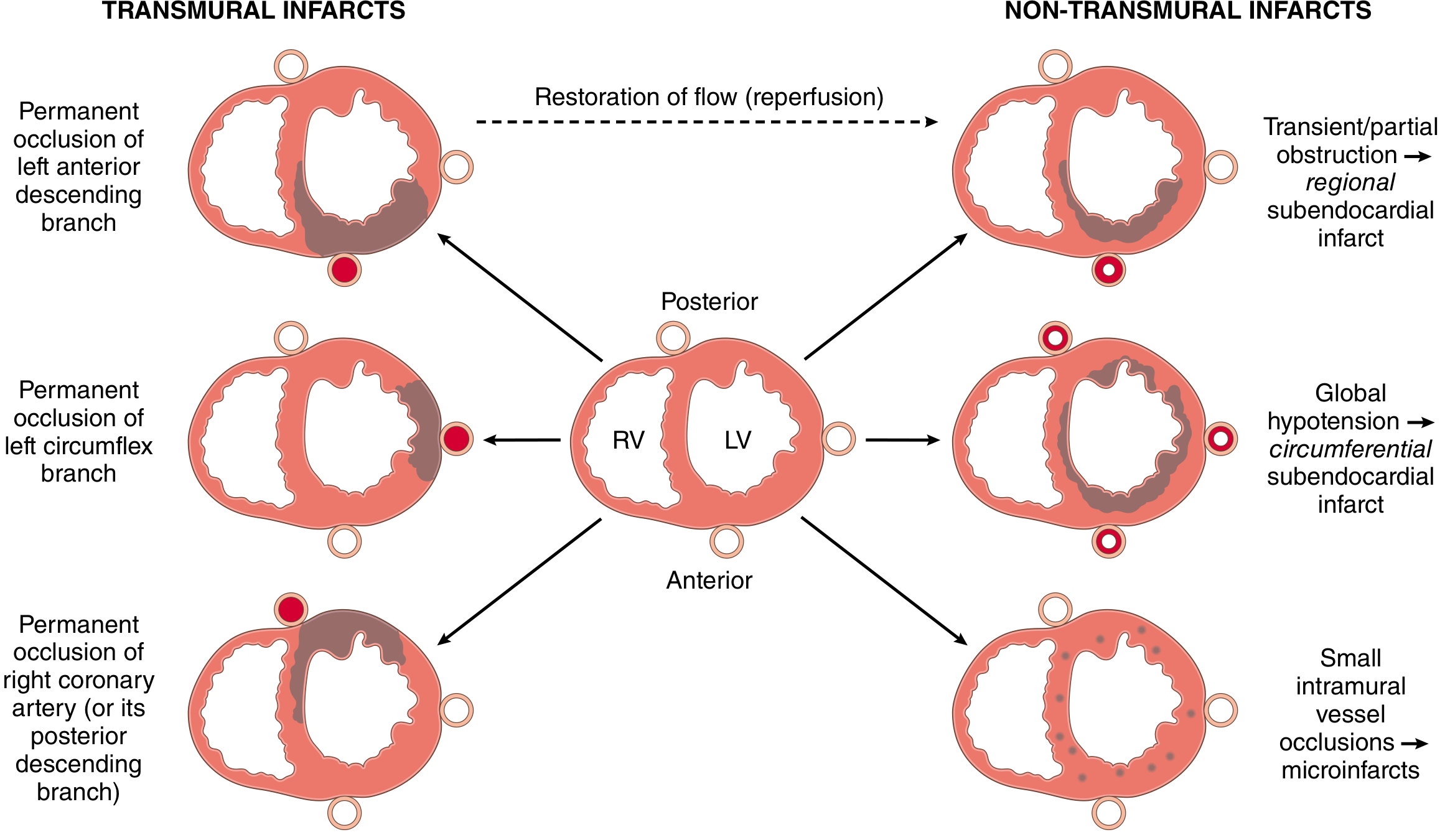
- LAD (40-50%): Anterior LV wall, anterior 2/3 of ventricular septum, apex
- RCA (30-40%): Inferior/posterior LV wall, posterior 1/3 of septum, posterior RV
- LCX (15-20%): Lateral LV wall
Gross Morphology (Table 12.5)
| Time after Onset | Gross Features |
|---|---|
| 0-12 hours | Usually none visible; TTC shows pale/unstained area |
| 12-24 hours | Dark mottling; pale with hemorrhagic periphery |
| 1-3 days | Mottled with pale yellow-tan necrotic center; hyperemia at border |
| 3-7 days | Hyperemic border; softening yellow-tan center (maximum softness ~day 5-7) |
| 7-10 days | Maximally yellow-tan, soft, depressed - maximum risk of rupture |
| 10-14 days | Red-gray depressed infarct borders; granulation tissue |
| 2-8 weeks | Gray-white scar tissue progressing from periphery inward |
| >2 months | Firm, contracted gray-white scar (complete healing) |
- A narrow rim (~0.1 mm) of viable subendocardial myocardium is preserved by diffusion from the ventricular lumen, even in transmural infarcts.
- STEMI (transmural): Full-thickness necrosis; associated with epicardial vessel occlusion
- NSTEMI (subendocardial): Necrosis confined to inner 1/3 to 1/2 of wall; associated with partial or transient occlusion, or global hypotension
Microscopic Examination
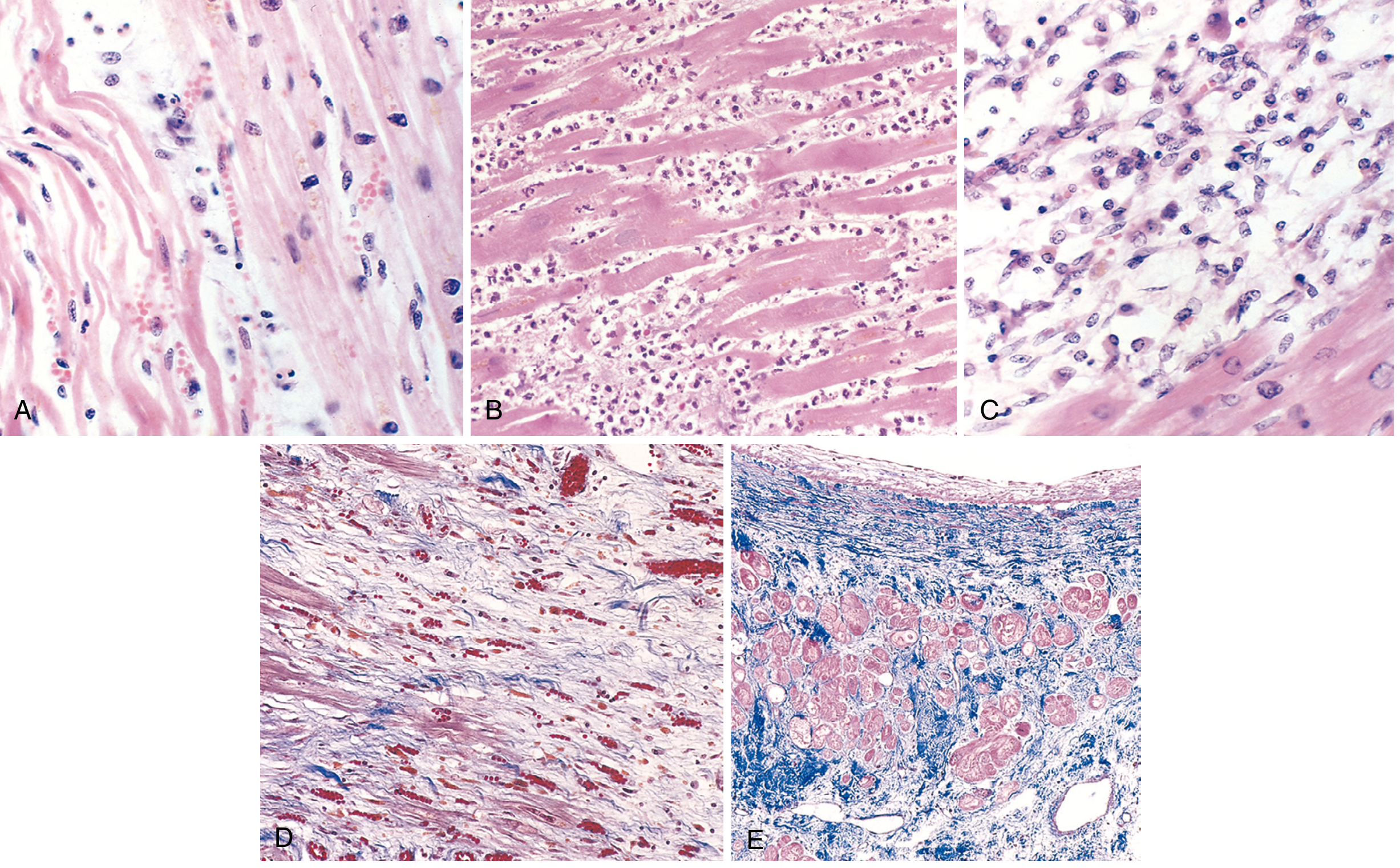
| Time | Histologic Features |
|---|---|
| 0-30 min | No visible change on light microscopy; electron microscopy shows myofibril relaxation, glycogen depletion, mitochondrial swelling |
| 1-4 hours | Earliest: waviness of fibers at border (due to non-contracting fibers being stretched); cell swelling |
| 4-12 hours | Coagulative necrosis begins; eosinophilic (hypereosinophilic) myocytes; nuclear pyknosis |
| 12-24 hours | Pyknotic nuclei; contraction bands; marginal neutrophil infiltration |
| 1-3 days | Coagulative necrosis with loss of nuclei and striations; heavy acute neutrophilic infiltrate (PMNs); interstitial edema |
| 3-7 days | Dead myofibers begin to disintegrate; neutrophils replaced by macrophages; beginning of phagocytosis |
| 7-10 days | Active phagocytosis by macrophages removing necrotic debris; early granulation tissue at margins; maximum softness and risk of rupture |
| 10-14 days | Well-established granulation tissue (proliferating capillaries, fibroblasts, loose collagen) |
| 2-8 weeks | Collagen deposition; progressive fibrosis; scar formation from periphery inward |
| >2 months | Dense collagenous scar (Masson trichrome = blue); residual myocytes show compensatory hypertrophy |
Diagnosis
- Prolonged (>30 minutes) crushing, squeezing, or stabbing substernal chest pain
- Radiation to left arm, jaw, or back
- Diaphoresis, nausea, vomiting
- Dyspnea (impaired contractility → pulmonary congestion)
- Rapid, weak pulse
- In ~25% of patients, onset is entirely asymptomatic (especially in diabetics due to neuropathy and in the elderly)
- STEMI: ST segment elevation in leads overlying the infarct (caused by rapid repolarization, decreased resting membrane potential, and delayed depolarization of infarcted fibers - all generating current flowing outward from the infarct); reciprocal ST depression in opposite leads; Q wave development after days-weeks
- NSTEMI: ST depression or T-wave inversions; no Q waves
- Non-Q-wave infarcts tend to be less severe but have high risk of reinfarction
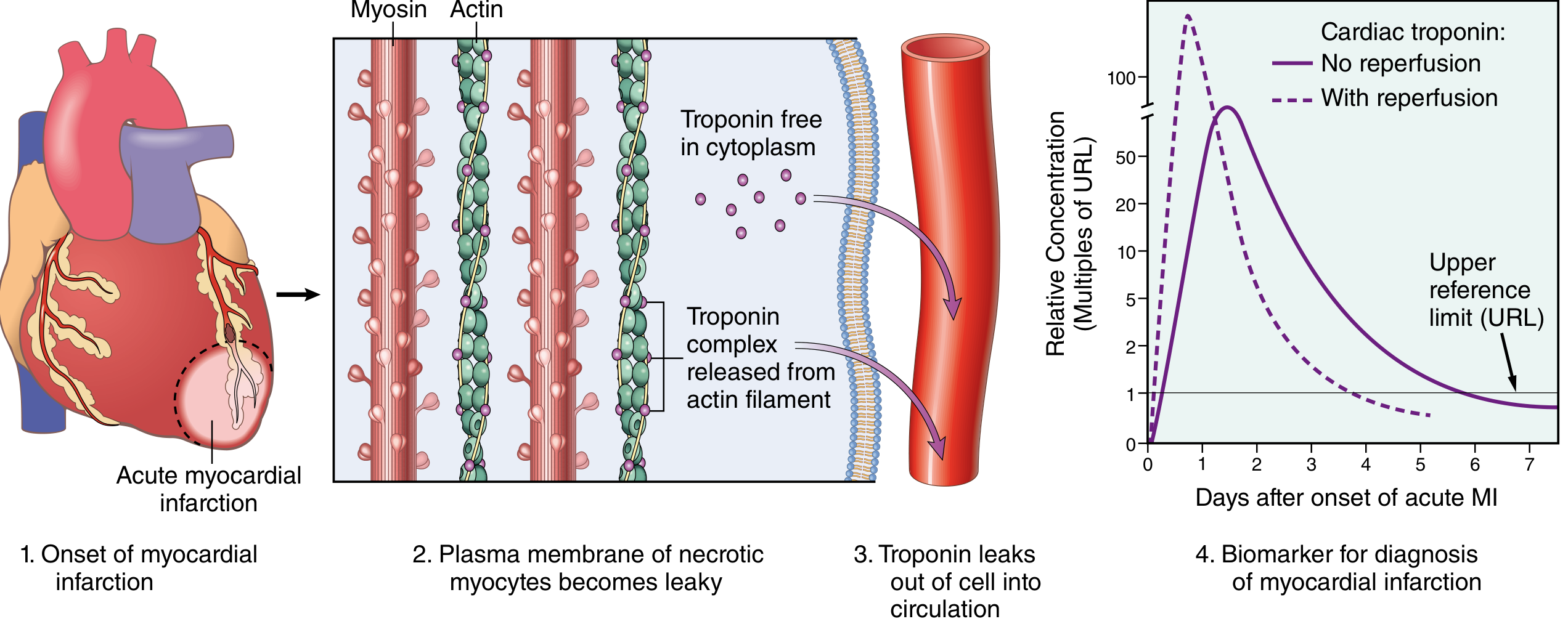
- Cardiac Troponin I & T (cTnI, cTnT): Most clinically useful; begin rising at 2-4 hours, peak at 24-48 hours, remain elevated for 7-10 days; with reperfusion, peak higher and earlier (washout effect)
- CK-MB: Rises by 4-6 hours, peaks 18-24 hours, normalizes by 72 hours - useful for detecting reinfarction
- Myoglobin: Earliest marker (within 1-2 hours) but not cardiac-specific
- LDH (LDH1 > LDH2): Historical; rises late, peaks at 3-6 days, not preferred today
- Echocardiography: regional wall motion abnormalities
- Coronary angiography: gold standard for identifying culprit lesion
- Radionuclide imaging, CT coronary angiography, MRI (late gadolinium enhancement for scar)
Treatment
- Oxygen - supplementation for hypoxic patients (SaO2 <90%)
- Nitrates - vasodilation, reversal of vasospasm; sublingual NTG initially
- Aspirin + P2Y12 inhibitors (clopidogrel, ticagrelor, prasugrel) - antiplatelet
- GPIIb/IIIa inhibitors - in selected high-risk PCI patients
- Anticoagulants - unfractionated heparin, LMWH, direct thrombin inhibitors, or factor Xa inhibitors (to prevent thrombus propagation)
- Beta-blockers - reduce myocardial O2 demand, reduce arrhythmia risk; give early unless contraindicated (heart failure, bradycardia, bronchospasm)
- Morphine - analgesia, reduces sympathetic activation
- Prompt reperfusion - the single most effective intervention (see below)
- ACE inhibitors / ARBs - reduce ventricular remodeling, especially post-STEMI
- Beta-blockers - ongoing cardioprotection
- Statins - stabilize plaques, reduce LDL
- Dual antiplatelet therapy for 12 months post-PCI
- Aldosterone antagonists (eplerenone/spironolactone) in EF <40%
Reperfusion & Reperfusion Injury
- Ischemia of 20-30 minutes → reversible injury → full recovery with reperfusion
- Longer ischemia → irreversible necrosis in affected zone
- But reperfusion can also salvage non-necrotic at-risk tissue
- Fibrinolysis (thrombolysis) - tPA, streptokinase
- Percutaneous coronary intervention (PCI) - preferred when available
- CABG (in selected cases)
- Mitochondrial dysfunction - ischemia alters mitochondrial membrane permeability; reperfusion causes swelling and outer membrane rupture, releasing pro-apoptotic contents
- Myocyte hypercontracture - intracellular Ca2+ accumulates during ischemia; reperfusion augments uncontrolled myofibril contraction → cytoskeletal damage and cell death (contraction band necrosis on microscopy)
- Oxidative stress (free radicals) - increased generation of reactive oxygen species (O2•-, H2O2, •OH, peroxynitrite) from reperfused endothelial cells, damaged parenchymal cells, and infiltrating leukocytes; compromised antioxidant defenses sensitize cells to further damage
- Calcium overload - influx through damaged sarcolemma and ROS-injured sarcoplasmic reticulum → mitochondrial permeability transition pore (mPTP) opens → ATP depletion → further injury
- Inflammation - "danger signals" from dead cells + cytokines from resident macrophages + increased adhesion molecule expression on hypoxic endothelium → neutrophil recruitment → additional tissue destruction
Intervention (PCI)
- The preferred reperfusion strategy for STEMI when available within 90 minutes of first medical contact (vs. 120 minutes of symptom onset)
- Balloon angioplasty + drug-eluting stent (DES) placement at culprit lesion
- Superior to fibrinolysis in reducing death, reinfarction, and stroke
- Indicated for multivessel disease, left main disease, or failed PCI
- Saphenous vein grafts or internal mammary artery grafts
- Invasive strategy (early angiography + PCI/CABG) vs. conservative strategy based on risk score (TIMI, GRACE)
- High-risk features (elevated troponins, hemodynamic instability, recurrent ischemia) favor early invasive approach
Complications of MI
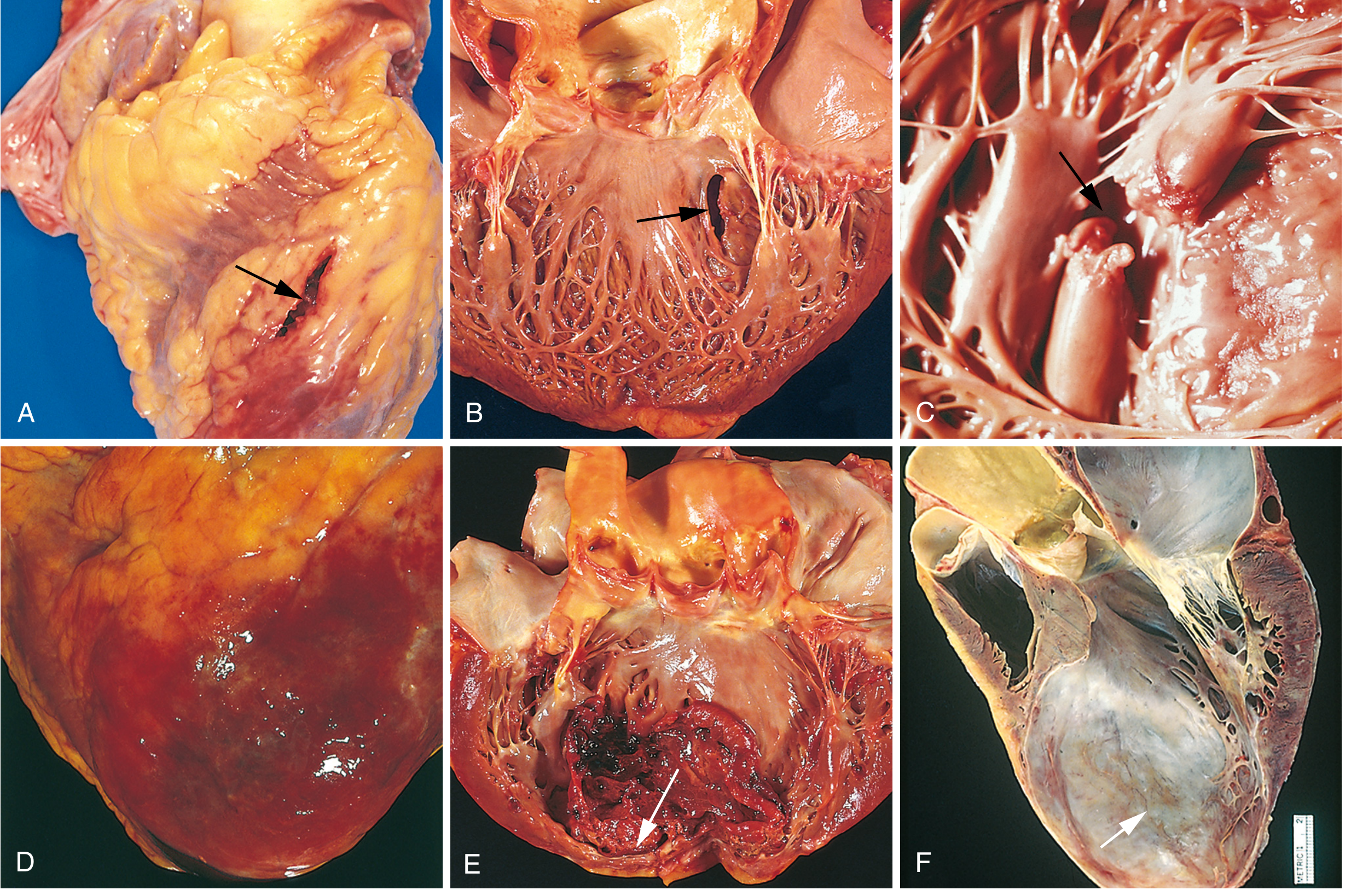
Early Complications (hours to days)
| Complication | Details |
|---|---|
| Arrhythmias | ~90% of patients develop some rhythm disturbance; risk highest in first hour; includes VF, VT, heart block, bradycardia, AF; most common cause of early death |
| Contractile dysfunction | LV pump failure proportional to infarct size; hypo-tension, pulmonary edema; cardiogenic shock in ~10% of transmural MI (when ≥40% LV affected) |
| Right ventricular infarction | From RCA occlusions; right heart failure → systemic venous pooling + systemic hypotension |
| Papillary muscle dysfunction | Ischemic dysfunction → mitral regurgitation (most common form of early MR); frank rupture is rare but catastrophic |
| Free wall rupture | 1-3% of MIs; peak risk 5-10 days (maximum softness); fatal hemopericardium + cardiac tamponade; more common with anterior transmural infarcts, first MI, elderly, hypertensive women |
| Ventricular septal rupture | Creates acute VSD with left-to-right shunt; loud pansystolic murmur; cardiogenic shock |
| Pericarditis (early) | Fibrinohemorrhagic pericarditis; 2-3 days post-MI; friction rub, anterior chest pain; transmural MIs only |
Late Complications (weeks to months)
| Complication | Details |
|---|---|
| Ventricular aneurysm | Large transmural anteroseptal infarct heals with thin fibrous wall; causes mural thrombus, arrhythmias, heart failure; does NOT rupture |
| Mural thrombus | Combination of stasis (poor contractility) + endocardial damage (thrombogenic) → thrombus in LV cavity → systemic embolism risk |
| Ventricular remodeling | Noninfarcted segments undergo hypertrophy and dilation; initially compensatory, then maladaptive; worsened by ventricular dilation and increased O2 demand; ACE inhibitors reduce this |
| Progressive heart failure (Chronic IHD) | Progressive functional decline from loss of viable myocardium + remodeling |
| Dressler Syndrome | See below |
| Post-MI mitral regurgitation (late) | From papillary muscle fibrosis/shortening or global ventricular dilation |
- Larger, transmural infarcts → higher probability of cardiogenic shock, arrhythmias, late CHF
- Anterior transmural: Higher risk of free-wall rupture, dilation, mural thrombi, aneurysm
- Posterior transmural: Higher risk of conduction blocks, RV involvement
- Subendocardial infarcts: Pericarditis, rupture, and aneurysm rarely occur
- Poor prognostic factors in STEMI: female sex, age >70, diabetes, previous MI
Dressler Syndrome (Post-MI Syndrome)
- Friction rub
- Typical ECG changes (saddle-shaped ST elevation, PR depression)
- Pleuritic chest pain, fever
- May develop pericardial effusion and even cardiac tamponade
- NSAIDs (first-line) for 2-3 weeks
- Colchicine - reduces recurrence
- Steroids - reserved for refractory cases; note: steroids should be avoided early post-MI as they can impair conversion of infarcted myocardium to scar (leading to greater wall thinning and rupture risk)
- Usually self-limited
Summary of Key Points
| Topic | Core Concept |
|---|---|
| Most common etiology | Atherosclerotic plaque disruption + thrombosis |
| Critical ischemia time | 20-40 min for irreversible injury; complete by 6-12 h |
| Earliest microscopic change | Wavy fibers + hypereosinophilia (4-12 h) |
| Grossly visible earliest | 12-24 h (TTC staining can show at <12 h) |
| Maximum rupture risk | Days 5-10 (maximum softness) |
| Best biomarker | Cardiac Troponin I/T (rises 2-4 h, stays 7-10 days) |
| Reperfusion goal | "Time is myocardium" - restore flow ASAP |
| Reperfusion injury mechanism | Free radicals + Ca2+ overload + inflammation + mitochondrial dysfunction |
| Reperfusion morphology | Contraction band necrosis + hemorrhagic infarct |
| Most common early complication | Arrhythmia (~90%) |
| Most common cause of early death | Ventricular fibrillation |
| Cardiogenic shock threshold | ≥40% LV destroyed |
| Dressler syndrome timing | 1 week to months post-MI; autoimmune |
- Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th ed., Chapter 12 (pp. 510-519)
- Robbins & Kumar Basic Pathology, Chapter 4 (Ischemic Heart Disease)
- Mulholland & Greenfield's Surgery, 7th ed., Chapter on Pericardial Disease
Rheumatic fever : definition, epidemiology, pathogenesis , acute rheumatic fever, jones criteria, major &minor criteria, essential criteria, rheumatic carditis- ache vs chronic, fibrinous pericarditis, aschoff bodies, anitschkow cells , types of vegetation, arthritis in rheumatic fever, sydenham chorea, subcutaneous nodules, erythema marginatum, complications , treatment, diagnosis
Rheumatic Fever - Comprehensive Review
Definition
Epidemiology
- Age: Most common in children 5-15 years; rare before age 3, uncommon after 15. Carditis is more severe in younger children; arthritis predominates in adults (20% of first attacks).
- Global burden: ~34 million people worldwide live with RHD. In developing nations, ARF remains a leading cause of childhood mortality.
- Incidence: ARF incidence of 2-14 cases/100,000 children per year in endemic areas; rare in high-income countries due to antibiotic use and improved living conditions.
- RHD peaks in adults aged 25-34 years.
- Latent period: Symptoms begin approximately 2-3 weeks after streptococcal pharyngitis (average 18.6 days); Sydenham chorea follows a longer latency of 4-8 weeks.
- Attack rate: ARF develops in only ~1.6-2.5% of patients with untreated streptococcal pharyngitis, confirming host genetic susceptibility.
- Sex: Chorea is more common in females. Genetic susceptibility shows 44% concordance in monozygotic twins vs. 12% in dizygotic twins.
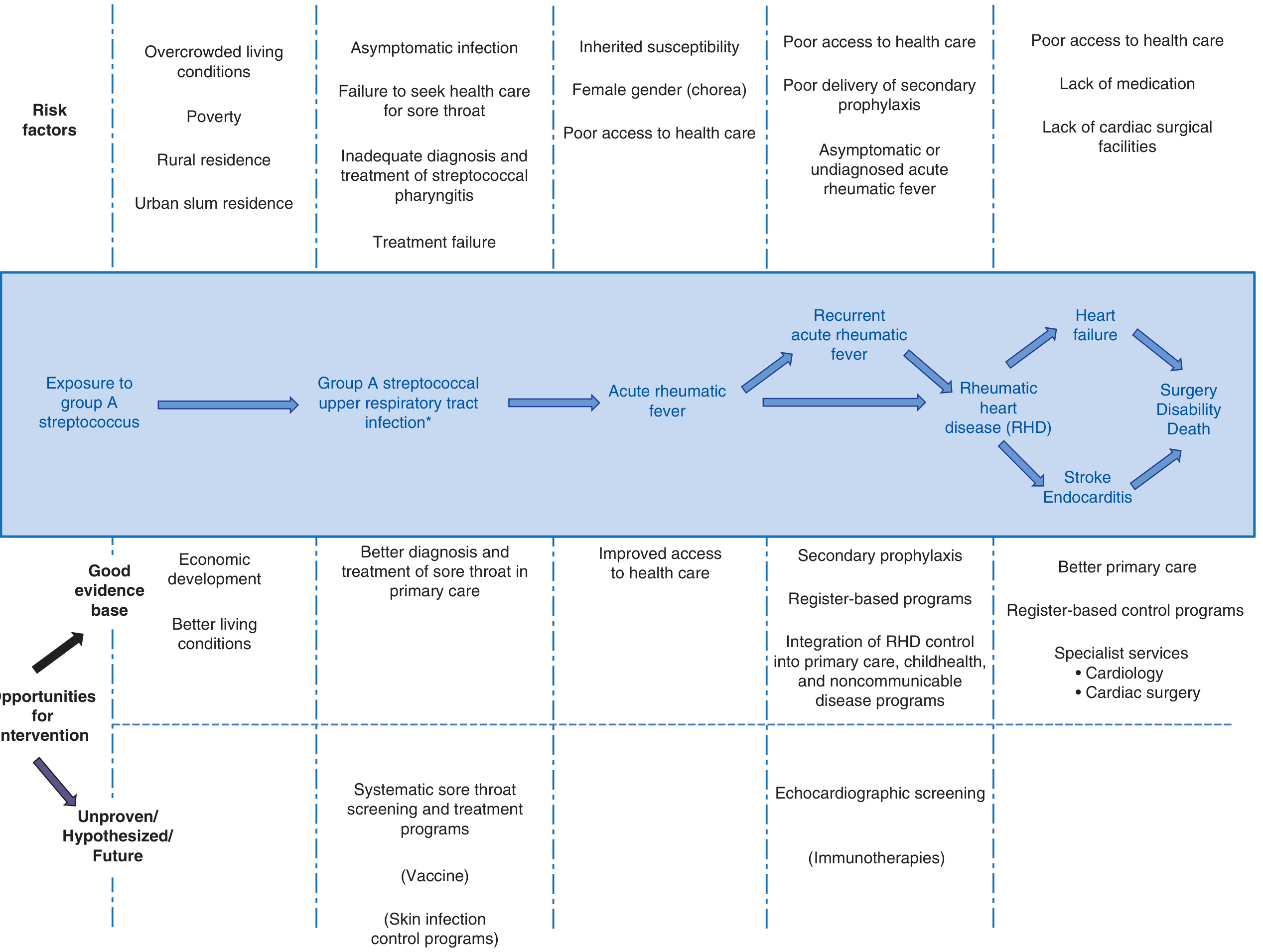
Pathogenesis
Organism Factors
Host Factors
- Only ~3-6% of any population is susceptible - genetic factors are key.
- HLA class II alleles, polymorphisms in TNF, mannose-binding lectin, IGHV4-61*02 allele, complement factor H, and multiple HLA-DQ loci are implicated.
Molecular Mimicry (Core Mechanism)
- GAS M protein antigens are presented by innate immune antigen-presenting cells to T cells.
- This activates both humoral (antibody) and cellular (T cell) immunity.
- Cross-reactive antibodies (targeting M protein epitopes) also bind to endothelial cells on heart valves, activating adhesion molecule VCAM-1.
- Activated lymphocytes are recruited and lyse endothelial cells (via complement).
- Damaged endothelium releases peptides (laminin, keratin, tropomyosin) that activate cross-reactive T cells.
- These T cells invade the heart, amplifying damage and causing "epitope spreading."
- Cytokine production by stimulated T cells leads to macrophage activation → Aschoff bodies form.
Acute Rheumatic Fever - Clinical Features
- Fever - high grade (≥39°C) in most cases; may be absent in pure chorea
- Migratory polyarthritis (most common and earliest major symptom)
- Carditis (pancarditis)
- Sydenham chorea
- Erythema marginatum
- Subcutaneous nodules
- Elevated acute-phase reactants (ESR, CRP)
Jones Criteria (2015 AHA Revision)
Essential Criterion (for ALL populations)
- Pure Sydenham chorea (may occur months after infection)
- Low-grade/subclinical carditis presenting late
- Positive throat culture for GAS or positive rapid streptococcal antigen test
- Elevated or rising streptococcal antibody titers - most commonly Anti-Streptolysin O (ASO) and Anti-DNase B (ADB)
Diagnosis Rules
| Diagnosis | Criteria Needed |
|---|---|
| Initial ARF | 2 major manifestations OR 1 major + 2 minor manifestations |
| Recurrent ARF | 2 major OR 1 major + 2 minor OR 3 minor manifestations |
Major and Minor Criteria by Population Risk
| Low-Risk Population | Moderate/High-Risk Population | |
|---|---|---|
| MAJOR CRITERIA | ||
| Carditis | Clinical or subclinical (echo) | Clinical or subclinical (echo) |
| Arthritis | Polyarthritis only | Polyarthritis OR monoarthritis OR polyarthralgia |
| Chorea | Sydenham chorea | Sydenham chorea |
| Subcutaneous nodules | Yes | Yes |
| Erythema marginatum | Yes | Yes |
| MINOR CRITERIA | ||
| Arthralgia | Polyarthralgia | Monoarthralgia |
| Fever | ≥38.5°C | ≥38°C |
| ESR/CRP | ESR ≥60 mm/h and/or CRP ≥3 mg/dL | ESR ≥30 mm/h and/or CRP ≥3 mg/dL |
| ECG | Prolonged PR interval (if carditis is NOT a major criterion) | Prolonged PR interval (if carditis is NOT a major criterion) |
Note: Arthralgia cannot be used as a minor criterion if arthritis is being used as a major criterion. Similarly, prolonged PR interval cannot be used as a minor criterion if carditis is a major criterion.
Rheumatic Carditis
Acute Rheumatic Carditis
1. Pericarditis
- Fibrinous pericarditis (fibrinohemorrhagic exudate)
- Manifests clinically in ~10% of patients as pleuritic chest pain and pericardial friction rub
- Pericardial effusion may be present; cardiac tamponade is rare
- Generally resolves without sequelae
2. Myocarditis
- Scattered Aschoff bodies in the interstitial connective tissue
- May cause cardiac dilation and functional mitral insufficiency
- Conduction system involvement → P-R interval prolongation (1st degree AV block); rarely higher-degree block
- Softening of the first heart sound
3. Endocarditis (Valvulitis) - Most Important
- Fibrinoid necrosis and fibrin deposition along the lines of closure of valve leaflets
- Forms 1-2 mm warty vegetations (verrucae) - see below
- Mitral valve involved in almost ALL cases of carditis
- Tricuspid valve frequently affected but rarely in a meaningful manner
- Aortic valve involved in 20-30% of cases
- Mitral regurgitation is the most common acute valvular lesion (from valvular inflammation, deformity, annular dilation, chordal elongation)
- Carey-Coombs murmur = low-pitched, apical, mid-diastolic flow murmur (due to acute mitral valvulitis)
- Heart failure occurs in 5-10% of first ARF episodes; more frequent with recurrences
Aschoff Bodies (Pathognomonic)
- Central zone of fibrinoid necrosis
- Surrounding lymphocytes (primarily T cells)
- Scattered plasma cells
- Anitschkow cells (pathognomonic macrophages)
- Plump, activated macrophages
- Abundant cytoplasm
- Nuclei with chromatin centrally condensed into a slender, wavy ribbon (the "caterpillar" appearance in longitudinal section; "owl-eye" in cross-section)
- Found in any layer of the heart during acute RF → evidence of pancarditis
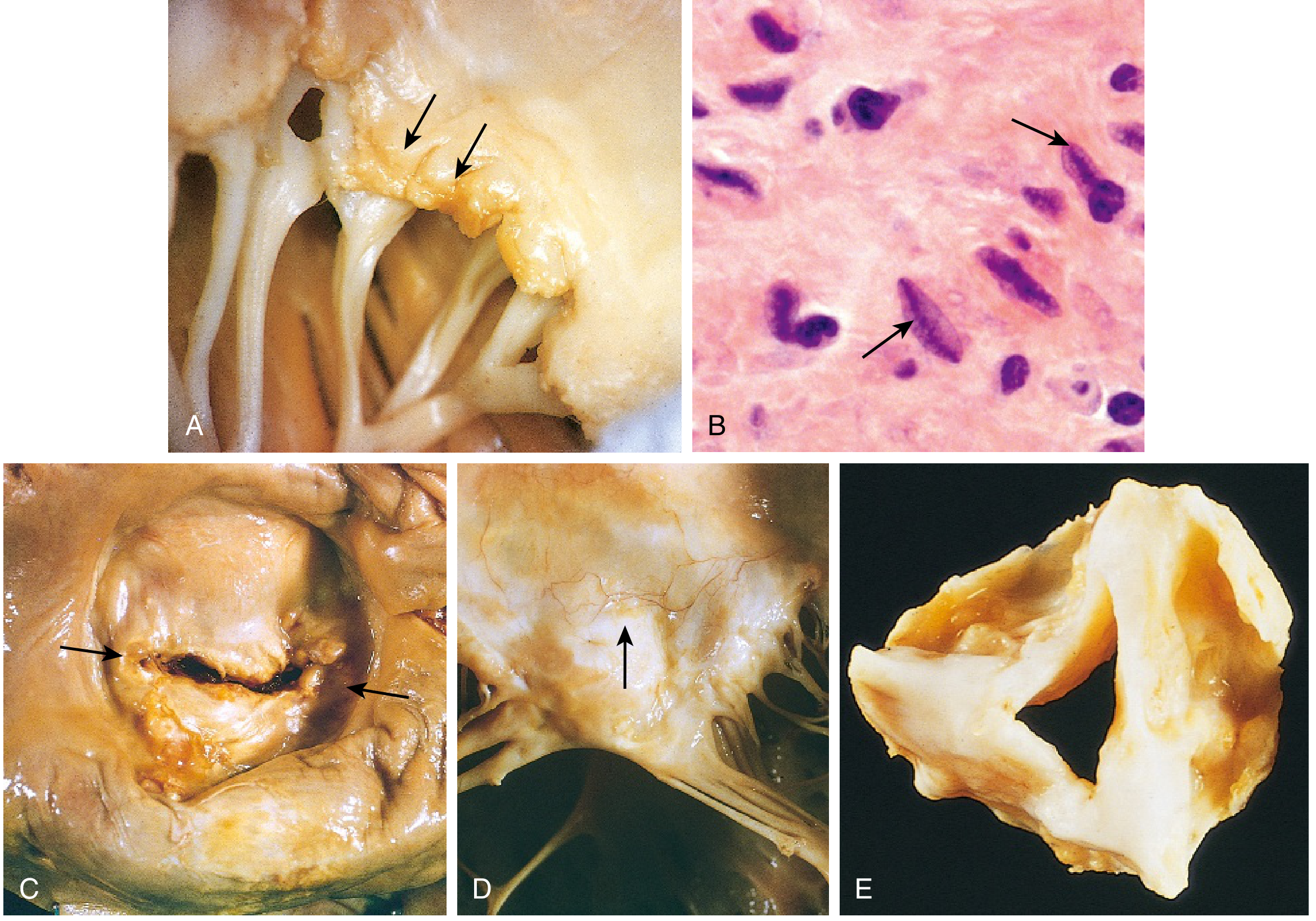
Types of Vegetation in Valvular Disease (Comparison)
| Type | Disease | Characteristics |
|---|---|---|
| Verrucae (rheumatic) | Acute Rheumatic Fever | Small (1-2 mm), warty, along the line of closure of valve leaflets; sterile; do NOT embolize; on the atrial surface of AV valves |
| Vegetations of infective endocarditis | Bacterial IE | Large, irregular, bulky, friable; can embolize; associated with valve destruction; on any surface |
| Libman-Sacks vegetations | SLE | Small, irregular; on both surfaces of valves (atrial AND ventricular); sterile; associated with antiphospholipid syndrome |
| Marantic (non-bacterial thrombotic) endocarditis | Debilitating illness, malignancy | Small, sterile; along line of closure; non-inflammatory; may embolize |
Chronic Rheumatic Heart Disease
- Aschoff bodies are replaced by fibrous scar (rarely seen in chronic disease)
- Valve cusps and leaflets become permanently thickened and retracted
- Commissural fusion - fibrous bridging across valvular commissures
- Shortening and thickening of chordae tendineae, with fusion
- Calcification → "fishmouth" or "buttonhole" stenoses (classic mitral stenosis appearance)
- Microscopy: neovascularization and diffuse fibrosis obliterating normal leaflet architecture
- Mitral valve alone: 70% of cases (most common acquired cause of mitral stenosis worldwide)
- Combined mitral + aortic: 25% of cases
- Tricuspid: less frequently and less severely involved
- Pulmonic valve: almost always spared
- Left atrial dilation (pressure overload from mitral stenosis) → atrial fibrillation
- AF + dilation → mural thrombus → systemic embolism (stroke)
- Pulmonary hypertension → right ventricular hypertrophy and failure
- Infective endocarditis (scarred, deformed valves are predisposed)
- Cardiac hypertrophy and dilation, CHF
People with RHD are often asymptomatic for years. In low-incidence countries, 20-40 years pass before surgery is required; in high-incidence countries, mitral stenosis can develop much more rapidly.
Major Manifestations - Detailed
1. Arthritis in Rheumatic Fever
- Most common (and often earliest) major manifestation of ARF
- Incidence increases with age: almost 100% in young adults, 82% in teenagers, 66% in children
- Classic pattern: migratory (flitting) polyarthritis - one large joint after another becomes painful and swollen for a few days, then spontaneously resolves, affecting the next joint
- Also seen: additive pattern (especially in adults)
- Joints affected most: knees (76%), ankles (50%), elbows and wrists (12-15%); less commonly shoulders, phalangeal, lumbosacral, cervical
- Pain is disproportionate to physical findings - exquisitely tender joints
- Sterile synovial fluid (inflammatory)
- No radiographic destruction - fully resolves without residual disability
- Responds dramatically to NSAIDs/salicylates ("aspirin test")
- Jaccoud's arthropathy (chronic post-RF arthropathy with reversible deformities) can occur after recurrent articular episodes
Arthritis typically lasts only a few days per joint and rarely more than 1 week per joint per ARF attack. In untreated patients, 6-16 joints are affected.
2. Sydenham Chorea (St. Vitus Dance)
- Involuntary, purposeless, non-repetitive choreiform movements
- Long latent period (4-8 weeks after GAS infection; sometimes months)
- More common in females and younger children
- Affects particularly the head (darting tongue movements) and upper limbs
- May be generalized or restricted to one side (hemichorea)
- Associated emotional lability and obsessive-compulsive traits
- Chorea usually resolves in 6 weeks but may take up to 6 months
- May occur in absence of other ARF manifestations (does not require positive streptococcal serology for diagnosis)
- >50% of patients presenting with pure chorea will have carditis - echocardiography is mandatory
- In severe cases: unable to perform activities of daily living
3. Subcutaneous Nodules
- Painless, small (0.5-2 cm), firm, mobile lumps
- Located beneath the skin overlying bony prominences
- Sites: hands, feet, elbows, occiput, occasionally vertebrae
- Delayed manifestation - appear 2-3 weeks after onset of disease
- Last only a few days to 3 weeks
- Strongly associated with carditis (rarely occur without carditis)
- Histologically resemble Aschoff bodies (fibrinoid necrosis with mononuclear infiltrate)
4. Erythema Marginatum
- Classic skin rash of ARF (but quite rare/evanescent)
- Pink macules that clear centrally, leaving a serpiginous, spreading, advancing edge
- Rash is evanescent - appears and disappears before the examiner's eyes
- Location: trunk, sometimes limbs; almost never on the face
- Non-pruritic
- May come and go with fever
- More common in children
5. Carditis (summarized above)
Fibrinous Pericarditis in Rheumatic Fever
- A fibrinohemorrhagic exudate coats the epicardial surface
- Grossly: rough, shaggy "bread-and-butter" appearance of epicardium (exudate on both surfaces sticking together)
- Microscopically: fibrin strands on pericardial surface with underlying inflammatory infiltrate
- Generally resolves without sequelae in RF (unlike other causes of pericarditis)
- Clinically: anterior chest pain, pericardial friction rub; effusion possible; tamponade rare
- Does NOT usually progress to constrictive pericarditis (unlike bacterial or tuberculous pericarditis)
Diagnosis
Clinical Diagnosis
- Evidence of preceding GAS infection (essential, except for pure chorea)
- Jones criteria (2015 revision, as above)
- Exclusion of other diagnoses
Investigations
- ECG - look for prolonged PR interval, ST/T changes
- Echocardiogram - to detect subclinical carditis, assess valves, determine severity
- CBC - neutrophilia, elevated WBC
- CRP (elevated)
- ESR (elevated)
- Streptococcal serology: ASO titer (Anti-streptolysin O) and Anti-DNase B (ADB) titers - one or both elevated in >95% of ARF; age-specific reference ranges apply; rising titer is more significant than single elevated value
- Throat swab culture for GAS
- Rapid streptococcal antigen test
- Synovial fluid analysis (sterile inflammatory fluid)
- Blood cultures
- Autoantibodies (ANA, ds-DNA, anti-CCP) to exclude other diagnoses
- Rises 1-3 weeks after GAS pharyngitis
- Peaks at 3-5 weeks
- Falls to pre-infection levels by 6-12 months
- May be falsely negative in chorea (occurs late) and skin infections
- If ASO negative but ARF suspected, check Anti-DNase B (remains elevated longer)
Treatment
1. Antibiotics (Eradication of GAS)
- Drug of choice: Penicillin
- Oral: Phenoxymethyl penicillin (penicillin V) 500 mg (250 mg for children ≤27 kg) PO twice daily × 10 days
- OR Amoxicillin 50 mg/kg (max 1 g) daily × 10 days
- OR single IM dose: Benzathine penicillin G 1.2 million units (600,000 units for children ≤27 kg)
- Penicillin allergy: Erythromycin or a narrow-spectrum cephalosporin
2. Anti-inflammatory Therapy
- Aspirin (salicylates) - first-line; 50-60 mg/kg/day up to 80-100 mg/kg/day (divided doses); typically given for 4-8 weeks, tapered as inflammation resolves
- NSAIDs (naproxen) - alternative
- Corticosteroids (prednisone 1-2 mg/kg/day, max 80 mg/day) - may be used for severe carditis with HF; taper over 2-4 weeks with overlapping aspirin to prevent rebound; no proven benefit on preventing long-term valvular damage
- No proven therapy alters the likelihood or severity of developing RHD
- Mild: symptomatic; aspirin/NSAIDs are of no value for chorea
- Moderate-severe: carbamazepine or valproic acid (anticonvulsants that reduce choreiform movements)
- Severe/refractory: IVIG (may lead to more rapid resolution; no benefit on carditis without chorea; not routinely recommended)
- Haloperidol or other antidopaminergic agents (second-line)
3. Heart Failure Management
- Bed rest
- Diuretics, ACE inhibitors/ARBs
- Digoxin (with caution in myocarditis)
- Temporary pacemaker if high-degree heart block with hemodynamic compromise
Secondary Prophylaxis (Prevention of Recurrence)
- Dose: 1.2 million units IM (600,000 units for children <27 kg) every 3-4 weeks
| Category | Duration |
|---|---|
| ARF without carditis | 5 years or until age 21, whichever is longer |
| ARF with mild/resolved carditis | 10 years or until age 21, whichever is longer |
| ARF with persistent mild-moderate valvular disease | 10 years or until age 40, whichever is longer |
| ARF with severe valvular disease or after valve surgery | Lifelong |
Complications
Acute Complications
- Severe carditis with acute heart failure (5-10% of first attacks; more with recurrences)
- High-degree heart block (rare, usually reversible)
- Pericardial tamponade (rare)
- Chordae tendineae rupture → acute HF requiring emergency surgery
- Chorea-related disability (self-limited but prolonged)
Chronic Complications
- Rheumatic heart disease - most significant long-term consequence
- Mitral stenosis (most common form; only acquired cause - "mitral stenosis is always rheumatic until proven otherwise")
- Mitral regurgitation (acute and chronic)
- Aortic stenosis/regurgitation
- Combined mitral + aortic disease (25%)
- Tricuspid involvement (less common)
- Atrial fibrillation (from left atrial dilation due to mitral stenosis)
- Systemic embolism/stroke (from mural thrombi in dilated left atrium + AF)
- Pulmonary hypertension → right heart failure
- Infective endocarditis (damaged valves are predisposed)
- Heart failure (long-term)
- Surgery/valve replacement requirement
Surgical repair or replacement (mitral valvuloplasty, valve replacement) has greatly improved long-term prognosis for patients with chronic RHD.
Summary Table
| Feature | Details |
|---|---|
| Causative organism | Group A beta-hemolytic Streptococcus (S. pyogenes) |
| Latency period | 2-3 weeks (chorea: 4-8 weeks) |
| Pathogenesis | Molecular mimicry - cross-reactive immunity (M protein vs myocardium/valves) |
| Pathognomonic lesion | Aschoff body (with Anitschkow/caterpillar cells) |
| Most common manifestation | Migratory polyarthritis |
| Most important manifestation | Carditis (determines long-term prognosis) |
| Valve most affected (acute) | Mitral (regurgitation) |
| Valve most affected (chronic) | Mitral (stenosis) - "fishmouth" deformity |
| Vegetation type | Verrucae (small, sterile, along line of closure) |
| Pathognomonic cell | Anitschkow cell ("caterpillar cell") |
| Diagnosis | Jones criteria + evidence of preceding GAS |
| Key serology | ASO titer + Anti-DNase B |
| Treatment: eradication | Benzathine penicillin G IM single dose |
| Treatment: arthritis/fever | Aspirin 50-80 mg/kg/day |
| Treatment: severe carditis | Corticosteroids |
| Treatment: chorea | Carbamazepine/valproate; IVIG if refractory |
| Secondary prophylaxis | Benzathine penicillin G 1.2 MU IM every 3-4 weeks |
Comparison of the four major forms of vegetative endocarditis make a table (add mnemonic if possible)
Comparison of the Four Major Forms of Vegetative Endocarditis
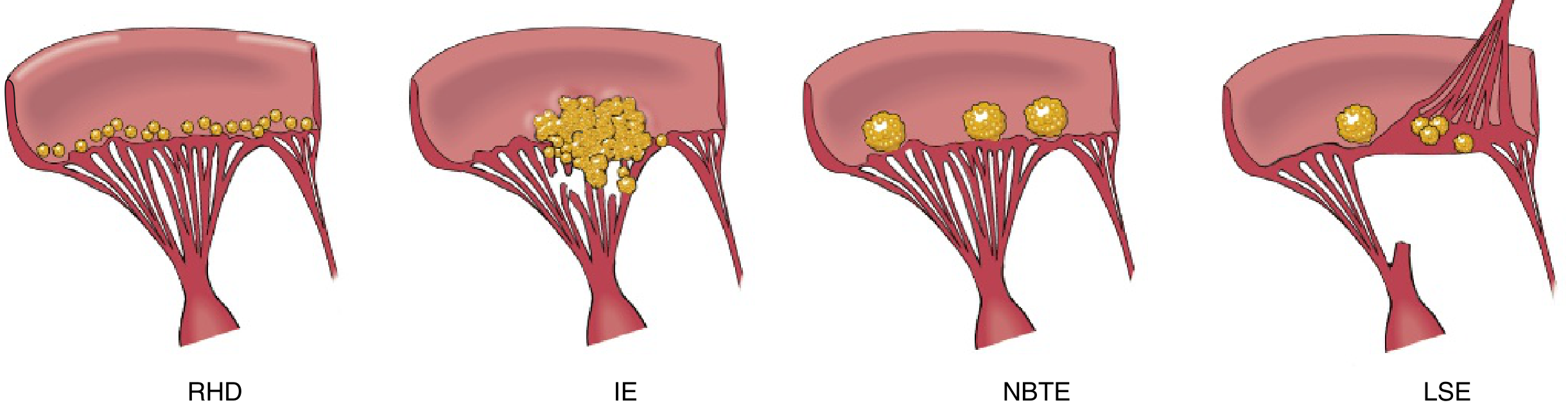
Mnemonic: "RINS" - Remember It's Not Simple
| Rheumatic | Infective | NBTE (Marantic) | SLE (Libman-Sacks) |
|---|
- Row = RHD verrucae line up in a row along closure line
- Bulky = IE has bulky, destructive, irregular masses
- Flat/sterile = NBTE is flat, sterile, loosely attached
- Both sides = LSE on both sides of valve (unique!)
Full Comparison Table
| Feature | Rheumatic Heart Disease (RHD) | Infective Endocarditis (IE) | NBTE / Marantic Endocarditis | Libman-Sacks Endocarditis (LSE) |
|---|---|---|---|---|
| Underlying disease | Acute rheumatic fever (post-GAS pharyngitis) | Bacteremia / septicemia | Malignancy (esp. mucinous adenocarcinoma), DIC, debilitating illness, hypercoagulable states | SLE; also antiphospholipid antibody syndrome |
| Pathogenesis | Molecular mimicry → immune-mediated valvulitis; fibrin deposits along closure line during healing | Microbial seeding of valve → local infection, destruction, thrombus formation | Hypercoagulable state → sterile platelet-fibrin thrombi on damaged or normal valves | Immune complex deposition + fibrinoid necrosis → sterile thrombotic lesions |
| Vegetation size | Small (1-2 mm) | Large, bulky, irregular (mm to cm) | Small to medium (1-5 mm) | Small (1-4 mm) |
| Vegetation appearance | Tiny, uniform, warty beads ("verrucae") in a neat row | Friable, bulky, irregular, cauliflower-like masses | Small, flat, irregular; loosely attached, may be multiple | Small, irregular, granular |
| Location on valve | Along the line of closure (atrial surface of AV valves, ventricular surface of semilunar valves) | Any surface of valve; often at base or tips; can destroy cusps | Free margin of valve leaflet; line of closure (atrial surface of AV valves) | BOTH surfaces of valve (atrial AND ventricular surface) - unique to LSE/NBTE-SLE |
| Valve most commonly affected | Mitral (70% alone); Mitral + Aortic (25%); Tricuspid sometimes; Pulmonic almost never | Aortic and Mitral (left-sided predominant); Tricuspid in IV drug users | Mitral (2/3); Aortic (1/3) | Mitral most common; any valve; can also involve chordae and mural endocardium |
| Sterile or infected? | Sterile | Infected (bacteria, fungi, etc.) | Sterile | Sterile |
| Valve destruction? | Minimal in acute phase; YES in chronic (fibrosis, stenosis years later) | YES - actively destructive; erosion, perforation, ring abscess | No (non-destructive) | Minimal/No in acute phase; may scar → deformity resembling chronic RHD |
| Commissural fusion? | YES (hallmark of chronic RHD) | No | No | No |
| Embolism? | Rare (verrucae are firmly adherent) | Common (friable → septic emboli → abscesses, mycotic aneurysms) | Common (nearly 50% experience systemic embolism - cerebral, renal, mesenteric) | Yes (can embolize → stroke) |
| Fever? | Present during acute RF | High fever (cardinal sign of IE) | Usually absent (from endocarditis itself) | Fever from underlying SLE |
| Microscopy / Histology | Fibrin + platelets on surface; underlying fibrinoid necrosis; Aschoff bodies in myocardium | Fibrin, platelets, microorganisms, PMNs; valve destruction | Fibrin + platelet thrombus; no organisms, no inflammation underneath | Fibrin + platelets; immune complexes + fibrinoid necrosis of underlying valve; inflammation |
| Organisms present? | No | YES (bacteria/fungi within vegetation) | No | No |
| Blood cultures | Negative | Positive (bacteremia) | Negative | Negative |
| Subsequent risk | Progressive valvular damage with recurrences | Can seed NBTE or damaged valves | Can become colonized → secondary IE | Valvular scarring similar to RHD |
| Treatment | Penicillin (prophylaxis); anti-inflammatories; valve surgery for chronic RHD | IV antibiotics for 4-6 weeks; valve surgery if needed | Anticoagulation (LMWH preferred); treat underlying cause | Treat underlying SLE; anticoagulation if antiphospholipid syndrome |
| Prognosis / Outcome | Chronic scarring → mitral stenosis/regurgitation years later | Fatal if untreated; mortality even with treatment | Clinically silent until embolism; associated with advanced malignancy (poor prognosis) | Generally associated with systemic SLE activity |
Memory Hook for Each Type
| Type | Hook |
|---|---|
| RHD | "Neat row of beads along the seam" - small, sterile, in a line, no destruction in acute phase |
| IE | "Bulky and bad" - large, infected, destructive, embolic, fever, positive cultures |
| NBTE | "Marantic = cancer/clot" - sterile, non-destructive, but embolic; think malignancy/DIC |
| LSE | "Lupus loves both sides" - only form with vegetations on BOTH surfaces of the valve |
Quick Reference: The "4 S's" of Vegetation Character
| Size | Sterile? | Destructive? | Special location | |
|---|---|---|---|---|
| RHD | Small (1-2mm) | Yes | No (acute) / Yes (chronic) | Line of closure only |
| IE | Large/bulky | No (infected) | Yes | Any surface |
| NBTE | Small-medium (1-5mm) | Yes | No | Line of closure / free margin |
| LSE | Small (1-4mm) | Yes | Minimal | Both surfaces |
Cardiomyopathy: definition, types For each type : causes, pathogenesis, gross and microscopy examination, diagnosis, special points, treatment Also make a table to differentiate between them (Add mnemonic if possible)
Cardiomyopathy - Comprehensive Review
Definition
- Primary: Principally confined to the myocardium
- Secondary: Cardiac manifestation of a systemic disorder
Important exclusions: myocardial dysfunction secondary to coronary artery disease, hypertension, valvular disease, and congenital heart disease are NOT classified as cardiomyopathies.
Types (Three Major Patterns)
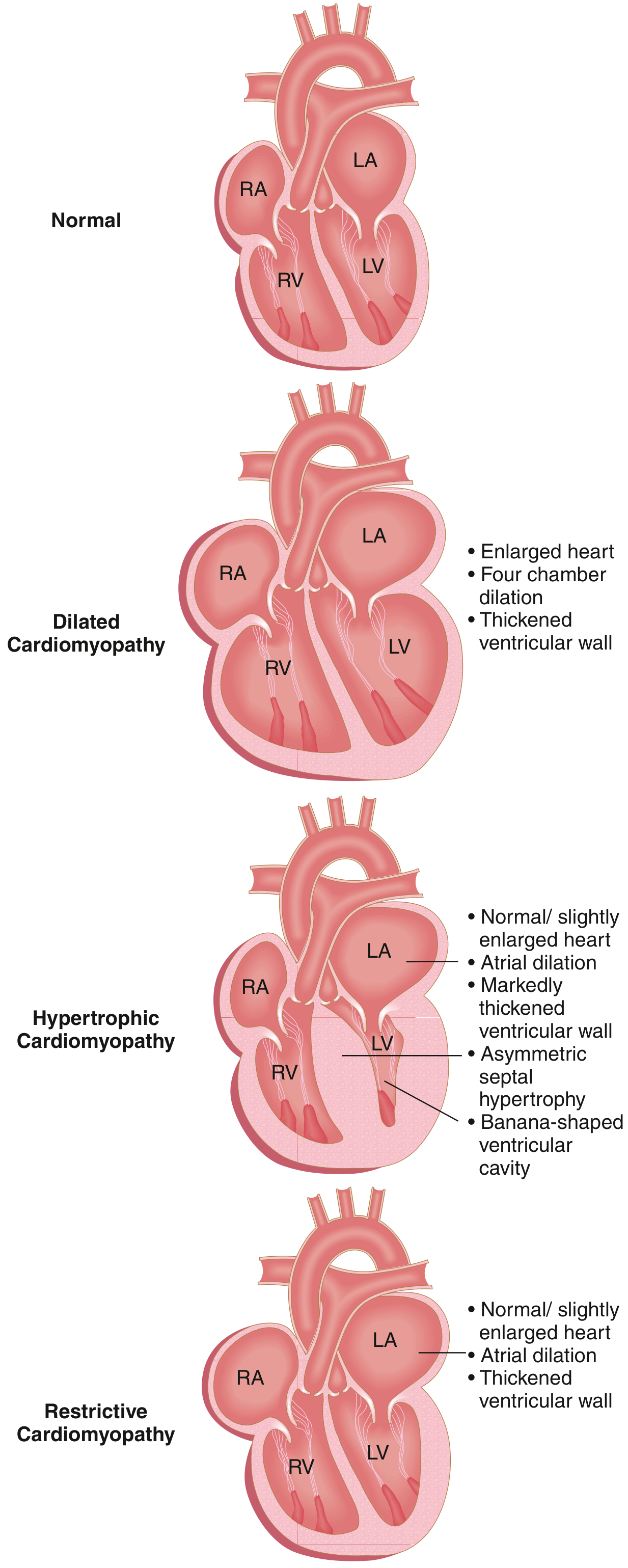
- Dilated Cardiomyopathy (DCM) - most common (90% of cases)
- Hypertrophic Cardiomyopathy (HCM)
- Restrictive Cardiomyopathy (RCM) - least common
- Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) - sometimes listed as a 4th type
Mnemonic: "D-H-R = Dilates, Hypertrophies, Restricts"
- DCM = heart gets Doughy and Dilated (can't squeeze)
- HCM = heart gets Huge and Hard (can't relax)
- RCM = heart gets Rigid and Restricted (can't fill)
1. Dilated Cardiomyopathy (DCM)
Definition
Causes / Etiology
- Idiopathic (most common, ~50%)
- Alcohol / toxic (alcohol + acetaldehyde direct toxicity; also cocaine, doxorubicin/anthracyclines, heavy metals)
- Myocarditis → viral (coxsackievirus B, parvovirus B19, HHV-6, enteroviruses, adenovirus, HIV)
- Anemia (chronic severe)
- Beritberi (thiamine/nutritional deficiency)
- Chaga's disease (Trypanosoma cruzi); also Hemochromatosis
- DNA mutations - Genetic (20-50% hereditary)
- Loss-of-function mutations in sarcomeric proteins or cytoskeletal linker proteins
- Titin (TTN) mutations - most common genetic cause
- β-myosin heavy chain, α-myosin heavy chain, cardiac troponin T
- Dystrophin mutations (X-linked DCM; also seen in Duchenne/Becker muscular dystrophy)
- Desmin (intermediate filament protein) and nuclear lamins A/C
- Peripartum cardiomyopathy (last trimester / first 6 months post-delivery)
- Sarcoidosis
- Thyroid disease (hypo- or hyperthyroid)
Pathogenesis
- Myocyte damage (via genetic, toxic, or infectious mechanisms)
- Loss of contractile function → compensatory ventricular dilation and hypertrophy
- Progressive pump failure (systolic dysfunction)
- Secondary mitral/tricuspid regurgitation (from annular dilation)
- Low cardiac output → neurohormonal activation (RAA, sympathetic) → fluid retention, more dilation → vicious cycle
Gross Morphology
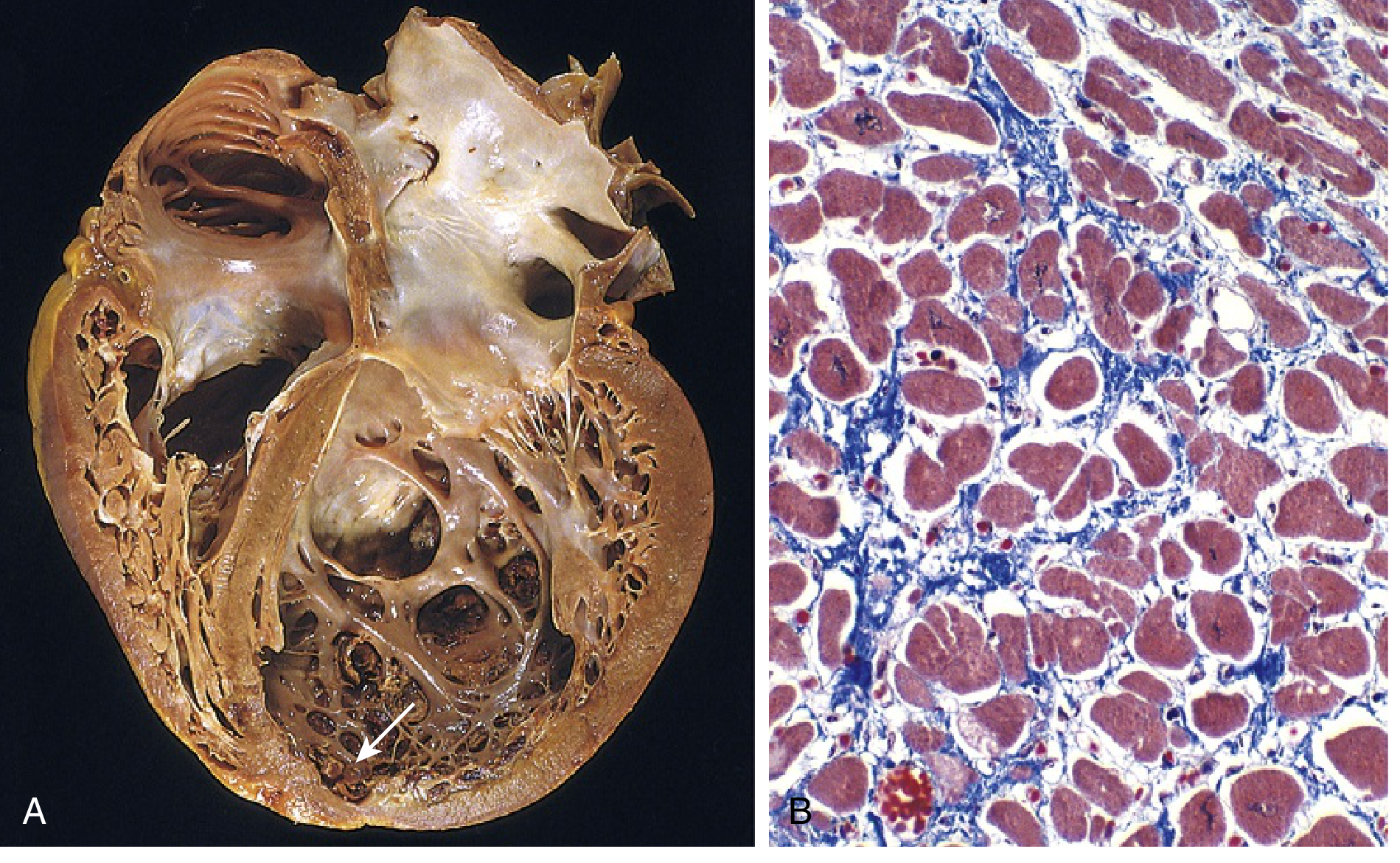
- Enlarged, heavy, flabby heart ("globoid" shape)
- 4-chamber dilation (especially left-sided)
- Ventricular wall: may be thickened, thinned, or normal
- Mural thrombi often present in dilated chambers (especially LV apex) → embolic risk
- Mitral and tricuspid valves may show functional regurgitation secondary to dilation
- Coronary arteries normal (no primary coronary disease)
Microscopy
- Myocyte hypertrophy (large, irregular nuclei - "boxcar nuclei")
- Interstitial fibrosis (variable, patchy or diffuse)
- Myocyte loss (dropouts replaced by fibrosis)
- No specific diagnostic finding - the picture is non-specific
- May see scattered inflammatory cells (if post-myocarditis)
Diagnosis
- Echo: Dilated LV/all chambers; reduced EF (<40%); regional wall motion abnormalities
- ECG: Non-specific ST/T changes; LBBB common; arrhythmias
- CXR: Cardiomegaly; pulmonary congestion
- BNP/NT-proBNP: Markedly elevated (heart failure marker)
- Troponin: May be mildly elevated in acute decompensation
- Endomyocardial biopsy: Rarely reveals specific diagnosis; useful if myocarditis or infiltrative disease suspected
- Cardiac MRI: Late gadolinium enhancement (fibrosis pattern); useful for etiology
- Genetic testing: For familial/young-onset DCM
Special Points
- Most common cardiomyopathy (90% of all cardiomyopathies)
- Peripartum DCM: develops in previously healthy women; may partially or fully recover post-partum
- Alcohol-related: partial/full recovery with abstinence
- Doxorubicin toxicity: dose-dependent; related to total cumulative dose; free radical mechanism
- Dystrophin mutations: X-linked; associated with Duchenne/Becker muscular dystrophy
Treatment
- Treat underlying cause (abstain from alcohol, antiviral, treat thyroid disease)
- Heart failure medications: ACE inhibitors/ARBs, beta-blockers, aldosterone antagonists (spironolactone/eplerenone), SGLT2 inhibitors (dapagliflozin)
- Diuretics: For fluid overload
- Anticoagulation: For mural thrombi or AF
- Antiarrhythmics / ICD: For ventricular arrhythmias; ICD if EF <35%
- CRT (cardiac resynchronization therapy): For LBBB + EF <35%
- Heart transplantation: End-stage refractory DCM
2. Hypertrophic Cardiomyopathy (HCM)
Definition
Causes / Etiology
- Genetic (primary/most common): Autosomal dominant; >400 causative missense mutations in sarcomeric protein genes
- β-myosin heavy chain (MYH7) - most common (35-40%)
- Myosin-binding protein C (MYBPC3) - second most common (20-25%)
- Cardiac troponin T (TNNT2), troponin I (TNNI3)
- α-tropomyosin, actin, titin, etc.
- All are GAIN-OF-FUNCTION mutations enhancing myofilament function → myocyte hypercontractility
Contrast with DCM: same genes (e.g., β-myosin) but loss-of-function in DCM vs. gain-of-function in HCM
- Secondary/Other causes:
- Friedreich's ataxia
- Glycogen storage diseases (Pompe, Fabry)
- Infants of diabetic mothers (transient neonatal HCM)
- Noonan syndrome
Pathogenesis
- Gain-of-function sarcomere mutations → myocyte hypercontractility and energy inefficiency
- Myocyte hypercontractility → cell death → replacement fibrosis
- Fibrosis + hypertrophy → diastolic stiffness (cannot relax after systole)
- Asymmetric hypertrophy → interventricular septum bulges into LVOT
- Systolic anterior motion (SAM) of mitral valve → LVOT obstruction (in ~1/3)
- LV cavity becomes small, elongated, "banana-shaped"
- High LV pressure + massive hypertrophy + compressed intramural vessels → subendocardial ischemia (even without CAD)
Gross Morphology
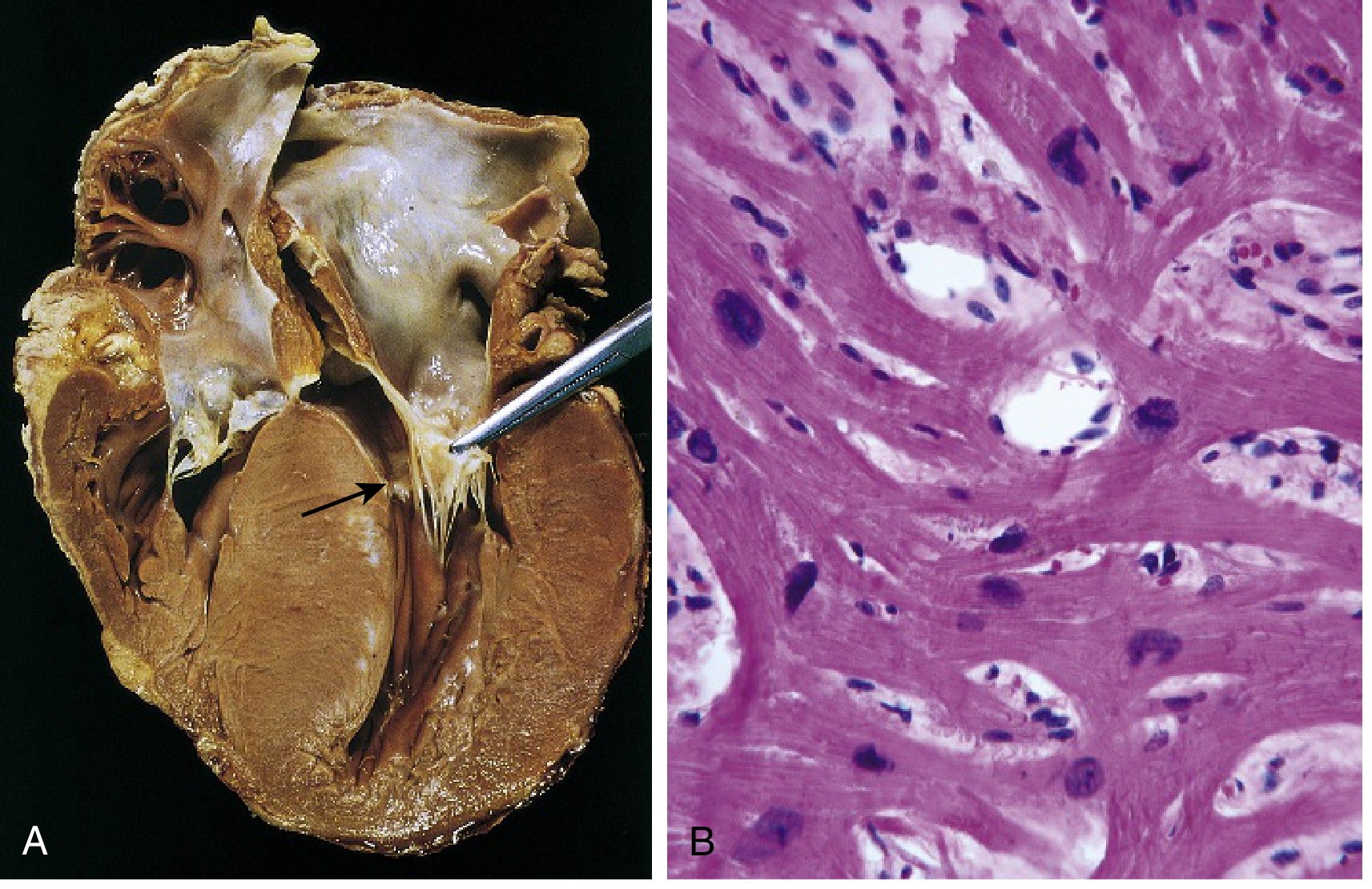
- Massive myocardial hypertrophy WITHOUT ventricular dilation
- Asymmetric septal hypertrophy (ASH) in 90% of cases (septum disproportionately thickened vs. LV free wall; septal:free wall ratio >1.3)
- Concentric hypertrophy in remaining 10%
- LV cavity: "banana-shaped" (compressed into elongated slit)
- Left atrium: dilated (from elevated LA pressure due to diastolic dysfunction)
- Fibrous endocardial plaque on LVOT (where anterior mitral leaflet contacts septum during SAM)
- Small-to-normal LV cavity; small or obliterated at end-systole
Microscopy
- Myocyte disarray (myofiber disarray): Haphazard, whirling arrangement of myocytes and myofibers (loss of parallel orientation) - pathognomonic of HCM
- Extreme myocyte hypertrophy with "boxcar" nuclei; exaggerated branching
- Interstitial fibrosis (replacement and reactive)
Diagnosis
- Echocardiography (gold standard):
- LV wall thickness ≥15 mm (in adults, unexplained)
- Asymmetric septal hypertrophy (septum:posterior wall ratio ≥1.3-1.5)
- Systolic anterior motion (SAM) of anterior mitral leaflet
- LVOT gradient ≥30 mmHg (obstructive HCM)
- Preserved/hyperdynamic EF (>50-80%)
- Diastolic dysfunction
- ECG: LVH, deep narrow Q-waves (from septal depolarization), T-wave inversions, LBBB, AF
- Cardiac MRI: Best for wall thickness quantification, fibrosis (late gadolinium)
- Genetic testing: Confirms diagnosis; guides family screening
- Auscultation: Harsh crescendo-decrescendo systolic ejection murmur at LLSB; increases with Valsalva/standing, decreases with squatting/hand-grip (classic dynamic maneuvers)
Special Points
- Most important cause of sudden cardiac death in young athletes (<35 years): HCM accounts for ~1/3 of SCD in athletes
- Autosomal dominant with variable expression (same mutation → different severity)
- Dynamic outflow obstruction: Valsalva, dehydration, standing all worsen obstruction
- Carey-Coombs murmur not applicable here; the LVOT murmur is the key
- Mitral regurgitation (from SAM) commonly accompanies obstruction
- Atrial fibrillation with mural thrombus is a major complication
Treatment
- Beta-blockers (first-line) - slow heart rate, promote filling, reduce LVOT gradient
- Non-dihydropyridine calcium channel blockers (verapamil, diltiazem) - same mechanism
- Disopyramide (negative inotrope) - reduce gradient
- Mavacamten (myosin inhibitor) - newer drug specifically for obstructive HCM
- Surgical septal myectomy (Morrow procedure) - gold standard for obstructive HCM
- Alcohol septal ablation - catheter-based controlled infarction of septal branches; alternative to surgery
- ICD: High-risk patients for SCD (family history SCD, massive hypertrophy ≥30 mm, unexplained syncope, NSVT, abnormal BP response to exercise)
- Anticoagulation for AF
- Avoid: Vasodilators, nitrates, diuretics (worsen outflow obstruction), digoxin
- Exercise restriction: Competitive sports discouraged
3. Restrictive Cardiomyopathy (RCM)
Definition
Causes / Etiology
- Radiation-induced fibrosis
- Amyloidosis (most common secondary cause in high-income countries)
- Idiopathic
- Sarcoidosis; Sphincolipidosis/Storage diseases (Gaucher, Fabry, mucopolysaccharidoses)
- Endomyocardial fibrosis (most common worldwide - tropical/Africa)
- Hemochromatosis (iron deposition)
- Loeffler endomyocarditis (hypereosinophilia)
- Metastatic tumors
- Carcinoid heart disease
- Most common cause in high-income countries
- AL amyloid (immunoglobulin light chains, in multiple myeloma/plasma cell dyscrasias)
- ATTR amyloid (transthyretin): Wild-type (senile) or hereditary (mutant transthyretin)
- 4% of African Americans carry V122I TTR mutation → 4-fold increased risk
- Cardiac amyloid → sparkling/granular appearance on echo; concentric LV thickening
- Classic cardiac echo: "sparkling" myocardium; low voltage on ECG despite thick walls
- Children and young adults in Africa and other tropical areas
- Worldwide most common form of restrictive cardiomyopathy
- Fibrosis from apex upward, eventually involving AV valves
- Linked to nutritional deficiency and/or parasitic infections (hypereosinophilia)
- Mural thrombi common
- Peripheral hypereosinophilia + eosinophilic tissue infiltrates
- Eosinophil major basic protein → endomyocardial necrosis → fibrosis + thrombus formation
- Associated with myeloid neoplasms with eosinophilia (FIP1L1-PDGFRA rearrangement)
Pathogenesis
- Infiltrative material or fibrosis stiffens the ventricular myocardium
- Impaired diastolic relaxation → elevated filling pressures
- Atria dilate (pressure overload from impaired ventricular filling)
- Systolic function initially preserved
- Progressive rise in venous pressures → right and left heart failure
Gross Morphology
- Ventricles: approximately normal size or slightly enlarged; cavities not dilated
- Firm myocardium
- Biatrial dilation (characteristic due to restricted ventricular filling)
- In amyloidosis: thick, rubbery ventricular walls with "waxy" texture
- In EMF: fibrous tissue lining of endocardium from apex upward
Microscopy
- Patchy or diffuse interstitial fibrosis (variable)
- Non-specific on routine biopsy; causes determined by special stains:
- Amyloid: Congo red staining → apple-green birefringence under polarized light
- Hemochromatosis: Iron stain (Prussian blue)
- Sarcoidosis: Non-caseating granulomas
- Endomyocardial biopsy often reveals a specific etiology (unlike DCM)
Diagnosis
- Echocardiography:
- Normal/near-normal LV size and EF
- Biatrial enlargement (prominent)
- Increased wall thickness (infiltrative causes)
- Diastolic dysfunction (abnormal E/e' ratio, restrictive filling pattern)
- "Sparkling" appearance in amyloidosis
- ECG: Low voltage relative to wall thickness (amyloid); conduction abnormalities
- Cardiac MRI: Late gadolinium enhancement patterns (diffuse subendocardial in amyloid; patchy in sarcoid)
- Nuclear imaging (Tc-99m pyrophosphate scan): Highly specific for ATTR amyloid
- Serum/urine protein electrophoresis + free light chains: For AL amyloid
- Endomyocardial biopsy: Congo red for amyloid, iron stains, granuloma detection - most useful in RCM
- Key clinical challenge: Must distinguish from constrictive pericarditis (similar physiology but different treatment!)
Special Points
- Least common of the three major cardiomyopathies
- Amyloidosis RCM key feature: "paradox of low voltage + thick walls" on ECG
- Must distinguish from constrictive pericarditis (which is treated with pericardiectomy)
- Constrictive pericarditis: calcified pericardium on imaging; normal myocardium; Kussmaul sign
- RCM: thick myocardium; normal pericardium; biopsy positive
- Endomyocardial fibrosis is most common worldwide but largely a disease of developing nations
Treatment
- Treat underlying cause:
- Amyloidosis: Tafamidis (TTR stabilizer) for ATTR amyloid; chemotherapy for AL amyloid
- Hemochromatosis: Phlebotomy/chelation
- Sarcoidosis: Corticosteroids
- Loeffler: Imatinib/tyrosine kinase inhibitors (if associated myeloid neoplasm)
- Diuretics: For fluid overload and elevated filling pressures (use cautiously)
- Rate control: For AF
- Anticoagulation: For AF or mural thrombi
- Heart transplant: End-stage disease
- EMF: Endocardiectomy with AV valve repair
4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Definition & Key Features
- Autosomal dominant disorder; prevalence 1:2000-1:5000
- Right-sided heart failure + rhythm disturbances → sudden cardiac death
- Mutation in desmosomal junction proteins (plakoglobin, desmoplakin, plakophilin, desmoglein, desmocollin)
- Also mutations in desmin (intermediate filament)
- Mechanism: desmosomal detachment (especially during strenuous exercise) → myocyte death → replacement by fat and fibrosis
Gross Morphology
- Right ventricular wall severely thinned due to myocyte replacement by fatty infiltration + fibrosis
- LV may also be involved to a lesser extent
Microscopy
- RV myocardium replaced by fibro-fatty tissue (fibrous + adipose tissue)
- Residual myocardial strands within fatty tissue
Diagnosis
- ECG: Epsilon waves (post-QRS notch in V1-V3); T-wave inversions in right precordial leads; RBBB morphology VT
- Echo/MRI: RV dilation, wall motion abnormalities, fatty infiltration
- Genetic testing for desmosomal mutations
- Task Force Criteria (major and minor) for diagnosis
Treatment
- ICD (high risk of SCD, especially in athletes)
- Antiarrhythmics (sotalol, amiodarone)
- Beta-blockers
- Exercise restriction (sports strongly discouraged)
- Catheter ablation for refractory VT
- Cardiac transplantation for refractory end-stage disease
Comparison Table
| Feature | DCM | HCM | RCM | ARVC |
|---|---|---|---|---|
| Mnemonic | Doughy/Dilated, can't Squeeze | Huge/Hard, can't Relax | Rigid, can't Fill | Right Fatty |
| Primary dysfunction | Systolic (pump failure) | Diastolic (filling failure) | Diastolic (filling failure) | Arrhythmic + RV failure |
| LV EF | <40% (reduced) | 50-80% (preserved/hyperdynamic) | 25-50% (preserved or mildly reduced) | Often preserved (RV affected) |
| Chamber size | All 4 chambers dilated | LV not dilated; LA dilated | LV not dilated; biatrial dilation | RV dilated; LV normal |
| Wall thickness | Normal, thin, or mildly thick | Markedly increased (asymmetric) | Increased (infiltrate) or normal | RV wall thinned (fat replacement) |
| Ventricular cavity | Dilated ("globoid") | Small, banana-shaped | Normal or slightly small | RV dilated |
| Myocardium feel | Flabby, soft | Firm, hypercontractile | Firm, stiff, rubbery | RV: soft/fatty |
| Classic cause | Idiopathic/alcohol/viral | Sarcomere mutation (β-MHC, MYBPC3) | Amyloidosis / idiopathic | Desmosomal mutation |
| Key gene mutation | Loss-of-function (titin, dystrophin) | Gain-of-function (β-MHC, MYBPC3) | N/A (usually secondary) | Desmosomal proteins (plakoglobin) |
| Histology hallmark | Myocyte hypertrophy + interstitial fibrosis (non-specific) | Myocyte disarray + extreme hypertrophy + interstitial fibrosis | Interstitial fibrosis ± specific infiltrate; Congo red+ (amyloid) | Fibro-fatty replacement of RV |
| Mural thrombus | Common (LV apex) | Less common | Can occur (EMF) | Less common |
| Outflow obstruction | No | Yes (in 1/3) - dynamic LVOT obstruction | No | No |
| Murmur | S3; MR murmur | Harsh systolic ejection murmur (increases with Valsalva) | S3/S4; typically quiet | Usually none |
| Special investigation | Echo; cardiac MRI; genetic testing | Echo (SAM + ASH); genetic testing | Endomyocardial biopsy; Congo red; Tc-99m PYP scan | ECG (epsilon wave); cardiac MRI |
| SCD risk | Moderate (VF) | High (esp. in young athletes) | Low | High (VF/VT in athletes) |
| Key treatment | ACE-I, beta-blockers, diuretics, ICD | Beta-blockers, verapamil, myectomy/alcohol ablation, ICD | Treat cause (tafamidis for ATTR); diuretics | ICD; exercise restriction |
| Avoid | (none specific) | Vasodilators, diuretics, nitrates (worsen obstruction) | Over-diuresis | Competitive sports |
| Looks like (mimic) | Ischemic/valvular/hypertensive heart disease | Aortic stenosis; hypertensive heart disease | Constrictive pericarditis | Myocarditis |
Summary Mnemonic: "D Flops, H Pops, R Stops, A Drops"
| Mnemonic | Meaning | |
|---|---|---|
| DCM | D FLOPS | Heart flops and dilates; pump flops |
| HCM | H POPS | Outflow pops shut (obstruction); athletes pop dead suddenly |
| RCM | R STOPS | Filling stops (restrictive); stiff wall; think Stops filling |
| ARVC | A DROPS | RV drops dead (arrhythmia); fatty infiltration makes RV drop |