Pathophysiology of pulmonary hypertension full details for md exam answer 10 marks
Reading File
Finding Sources
Searching PubMed
"pulmonary hypertension" AND pathophysiology
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive material from multiple authoritative textbooks. Let me compile the full, detailed MD exam answer:
Pathophysiology of Pulmonary Hypertension
Definition
Pulmonary hypertension (PH) is defined as a mean pulmonary artery pressure (mPAP) ≥ 25 mmHg at rest, measured by right heart catheterization. Normal mPAP is approximately 14 ± 3 mmHg. Pulmonary vascular resistance (PVR) is the key hemodynamic determinant.
WHO Classification (Etiological Framework)
The underlying cause dictates the pathophysiologic mechanism:
| Group | Category | Mechanism |
|---|---|---|
| 1 | Pulmonary Arterial Hypertension (PAH) | Vasoconstrictive + obliterative |
| 2 | Left heart disease | Passive (postcapillary) |
| 3 | Lung disease / hypoxia | Vasoconstrictive + obliterative |
| 4 | Chronic thromboembolic (CTEPH) | Obstructive |
| 5 | Unclear/multifactorial | Mixed |
Core Pathophysiologic Mechanisms
Regardless of the initial trigger, increased PVR in all forms of PH results from the collective effects of four processes:
- Sustained vasoconstriction
- Vascular remodeling
- In situ thrombosis
- Increased arterial wall stiffness
(Fishman's Pulmonary Diseases and Disorders)
1. Sustained Vasoconstriction
This is driven by a fundamental imbalance between vasoconstrictors and vasodilators at the level of the pulmonary endothelium.
Vasoconstrictors (Upregulated):
Endothelin-1 (ET-1):
- One of the most potent endogenous vasoconstrictors known
- Markedly increased in blood and lung tissue of PAH patients; correlates with disease severity
- Acts on ET-A receptors on pulmonary arterial smooth muscle cells (PASMCs) - causes potent vasoconstriction
- Also mitogenic for PASMCs and fibroblasts (promotes remodeling)
- ET-B receptors on smooth muscle: vasoconstriction + clearance of ET-1
- ET-B receptors on endothelium: paradoxically stimulate NO release (vasodilatory)
Thromboxane A2: Vasoconstrictor and platelet activator, increased in PAH.
Serotonin (5-HT): Acts via 5-HT transporter (5-HTT) to stimulate PASMC proliferation and vasoconstriction.
Angiotensin II: Promotes vasoconstriction and vascular smooth muscle proliferation.
Vasodilators (Downregulated):
Nitric Oxide (NO):
- Synthesized from L-arginine in endothelial cells by eNOS
- Diffuses into PASMCs, activates soluble guanylate cyclase (sGC) → increases cGMP → vasodilation and antiproliferation
- In PAH: eNOS expression and NO production are markedly reduced
- cGMP is also degraded by phosphodiesterase type 5 (PDE-5), which is upregulated in PH
Prostacyclin (PGI2):
- Synthesized from arachidonic acid in endothelial cells via cyclooxygenase
- Acts via IP receptor on PASMCs → increases cAMP → vasodilation and antiproliferation
- PGI2 synthase activity is markedly reduced in PAH
- Thromboxane A2/prostacyclin ratio is shifted toward vasoconstriction
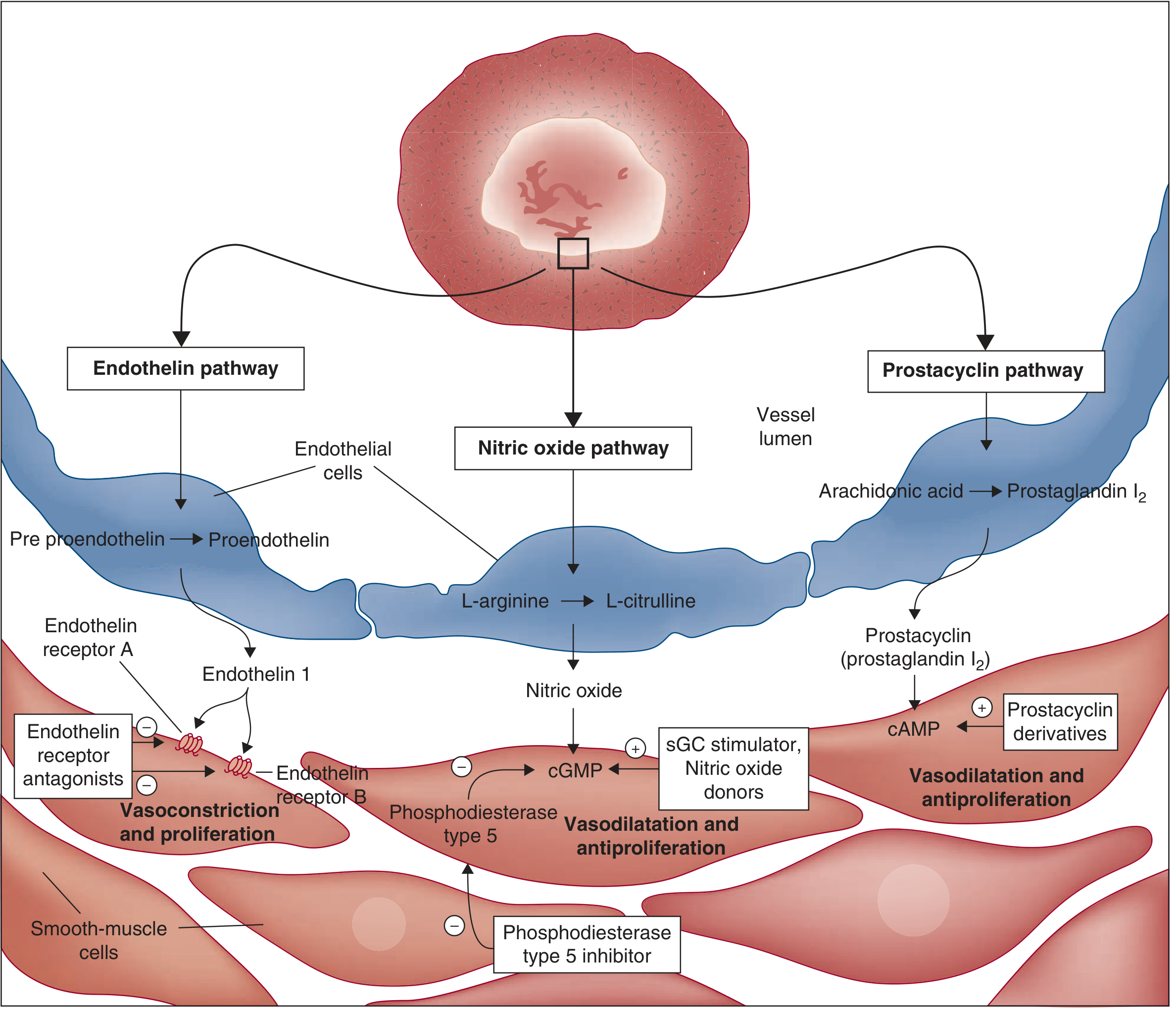
Fuster and Hurst's The Heart, 15th ed. - Three key pathways in PAH pathophysiology
Calcium Dysregulation in PASMCs:
- Rise in cytosolic free Ca²⁺ concentration in PASMCs is a major trigger for vasoconstriction
- Also a key stimulus for PASMC proliferation and migration
- K⁺ channel (Kv) downregulation → membrane depolarization → Ca²⁺ influx via voltage-dependent Ca²⁺ channels (VDCC)
- This represents an intrinsic property of PASMCs in response to hypoxia
Hypoxic Pulmonary Vasoconstriction (HPV):
- An adaptive mechanism to redirect blood from poorly ventilated to well-ventilated areas
- HPV is an intrinsic property of PASMCs (occurs even without endothelium)
- In chronic hypoxia: both sustained vasoconstriction AND structural remodeling occur within weeks
- Disrupted Ca²⁺ homeostasis is the principal mechanism
2. Vascular Remodeling
This is the most important structural change - it leads to irreversible narrowing of the pulmonary vascular bed.
The wall thickness is normally maintained by a balance between proliferation and apoptosis of:
- Fibroblasts
- Pulmonary arterial smooth muscle cells (PASMCs)
- Pulmonary arterial endothelial cells (PAECs)
When this balance tips toward proliferation, progressive wall thickening and luminal obliteration occur.
Structural changes include:
Medial Hypertrophy:
- Increase in number (hyperplasia) and size (hypertrophy) of PASMCs in the media
- Elevated PAP and sustained vasoconstriction perpetuate PASMC hypertrophy in a vicious cycle
- In advanced disease: medial atrophy, fibrosis, and paradoxical thinning with vessel dilation
Muscularization of Normally Non-muscular Vessels:
- Extension of PASMCs into small intra-acinar pulmonary vessels that are normally non-muscularized
- A prominent and early feature of PAH
- Seen in hypoxia-induced PH (indistinguishable histologically from IPAH)
Intimal Proliferation:
- Concentric laminar intimal thickening (onion-skin appearance) causes progressive luminal narrowing
- Results from proliferation of smooth muscle cells, myofibroblasts, and deposition of matrix proteins
Plexiform Lesions (characteristic of severe PAH):
- Aneurysmatic dilatations of small muscular arteries/arterioles
- Contain collections of proliferating endothelial cells, smooth muscle cells, myofibroblasts, and matrix proteins
- Partially or completely occlude the vessel lumen
- Found in IPAH, PAH associated with left-to-right cardiac shunts, HIV, liver cirrhosis, scleroderma
- In IPAH: endothelial cells proliferate in a monoclonal fashion (suggesting neoplastic-like behavior)
- In other PAH forms: polyclonal endothelial cell proliferation
Adventitial Changes:
- Adventitial fibroblast proliferation and increased collagen deposition → reduced arterial wall compliance and increased stiffness
(Fishman's Pulmonary Diseases and Disorders)
3. In Situ Thrombosis
- Monoclonal proliferation of PAECs + PASMC migration + accumulation of inflammatory cells, platelets, and progenitor cells → occlusion of smaller vessels
- Thrombosis occurs without a remote embolic source - reflects a local imbalance of pro- and anticoagulant forces
- Endothelial activation shifts the vessel from anticoagulant to procoagulant state (due to elevated shear stress)
- Decreased prostacyclin (antiplatelet, vasodilatory) and increased thromboxane A2 (proplatelet, vasoconstrictive) further promote thrombosis
- Von Willebrand factor and tissue plasminogen activator (tPA) changes impair fibrinolysis
4. Genetic and Molecular Mechanisms (BMPR2 Pathway)
This is most well-characterized in Group 1 (PAH):
BMPR2 Mutation:
- The first mutation identified in familial PAH
- BMPR2 = Bone Morphogenetic Protein Receptor type 2, a TGF-β receptor superfamily member
- Inactivating germline mutations in BMPR2 found in 75% of familial PAH and 25% of sporadic PAH
- BMPR2 is also downregulated in lungs without BMPR2 mutations (suggesting epigenetic silencing)
- Normal BMPR2 signaling: promotes apoptosis and inhibits proliferation of endothelial cells and VSMCs
- Loss of BMPR2 → haploinsufficiency → dysfunction and uncontrolled proliferation of endothelial cells and VSMCs → pulmonary vascular thickening and occlusion
Two-hit Hypothesis:
- Only 10-20% of BMPR2 mutation carriers develop disease
- A second hit (modifier genes, environmental trigger - e.g., hypoxia, inflammation, drugs, infection) is required
- This explains incomplete penetrance and the variable expressivity of familial PAH
Other genetic mutations (converging on BMPR2 pathway):
- ACVRL1 (activin receptor-like kinase 1) - associated with hereditary hemorrhagic telangiectasia
- ENG (endoglin) - also HHT-related
- SMAD1, SMAD4, SMAD9 - downstream transcription factors in the BMP pathway
- KCNK3 (potassium channel) - Ca²⁺ dysregulation
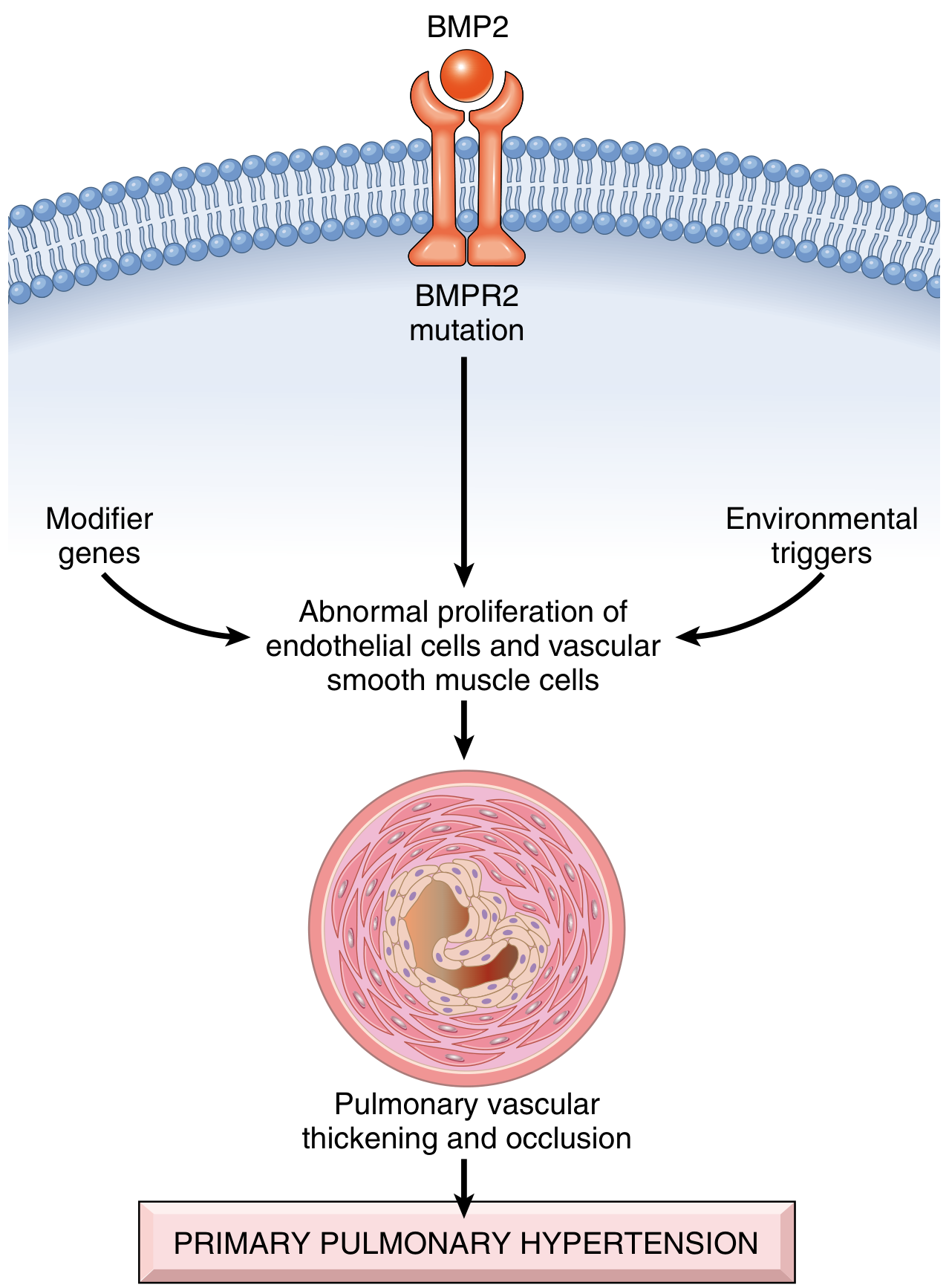
Robbins & Cotran Pathologic Basis of Disease - Pathogenesis of primary (idiopathic) pulmonary hypertension
5. Inflammation and Immune Mechanisms
- Inflammatory cell infiltrates (T and B lymphocytes, macrophages, mast cells, dendritic cells) are found in the plexiform lesions and surrounding vessels
- Elevated circulating cytokines: IL-1β, IL-6, IL-8, TNF-α, MCP-1
- Increased VEGF expression (particularly in plexiform lesions) promotes abnormal angioproliferation
- Growth factors implicated: PDGF, EGF, angiopoietin-1 (overexpressed in IPAH, correlates with severity)
- Regulatory T-cell dysfunction and autoantibodies suggest an autoimmune component
6. Pathogenic Mechanisms by Category (Fishman's Framework)
| Mechanism | Example |
|---|---|
| Passive | Obstruction to pulmonary venous outflow (mitral stenosis, left heart failure) - Group 2 |
| Hyperkinetic | High pulmonary blood flow (left-to-right shunts - VSD, ASD, PDA) - Group 1 |
| Obstructive | Pulmonary thromboembolic disease - Group 4 |
| Obliterative | Parenchymal proliferative disease (IPF, emphysema obliterate capillaries) - Group 3 |
| Vasoconstrictive | Chronic hypoxic vasoconstriction - Group 3 |
| Idiopathic | No discernible cause - Group 1 IPAH |
7. Right Ventricular Consequences (Cor Pulmonale)
The downstream effect of all the above mechanisms:
Right Ventricular Pressure Overload:
- Elevated PVR → elevated mean PAP → increased RV afterload
- RV hypertrophy (concentric) - compensatory, maintains cardiac output
- RV dilation - as compensation fails
- RV failure - the terminal event
Vicious Cycles:
- Elevated RV pressure → decreased coronary perfusion of RV wall → RV ischemia (supply-demand mismatch)
- RV dilation → interventricular septal shift toward LV (D-shaped septum on echo) → decreased LV preload → reduced systemic cardiac output → hypotension → further coronary ischemia
- RV failure → systemic venous hypertension → hepatic congestion, peripheral edema (clinical cor pulmonale)
Chest pain mechanism in PH: Either RV ischemia (inability to meet metabolic demands of hypertrophied RV) or compression of the left main coronary artery by the dilated main pulmonary artery trunk.
8. Group-Specific Pathophysiology Highlights
Group 2 (Left Heart Disease):
- Increased left atrial/PCWP pressure → passive back-transmission through pulmonary veins → pulmonary venous hypertension → reactive precapillary hypertension
- Transmits pulmonary venous pressure → medial hypertrophy of pulmonary arteries
Group 3 (Lung Disease/Hypoxia):
- Two mechanisms: HPV (vasoconstrictive) + parenchymal destruction/fibrosis (obliterative)
- COPD, ILD, sleep apnea all contribute through chronic hypoxemia
Group 4 (CTEPH):
- Acute thrombus → replaced by chronic intravascular scar
- Obstructs proximal vessels → elevated pressure → shunting of flow to remaining open vessels → secondary vascular remodeling of non-obstructed vessels (microvasculopathy)
- Approximately 4% of acute PE patients develop CTEPH
Summary Table: Key Mediators in PAH
| Mediator | Source | Effect | Net Result |
|---|---|---|---|
| Endothelin-1 | Endothelium | ET-A: vasoconstriction + proliferation | Increased PVR |
| Nitric Oxide (↓) | Endothelium via eNOS | Normally: cGMP ↑ → vasodilation | Decreased vasodilation |
| Prostacyclin (↓) | Endothelium via COX | Normally: cAMP ↑ → vasodilation | Decreased vasodilation |
| Thromboxane A2 (↑) | Platelets | Vasoconstriction + platelet aggregation | Thrombosis + ↑ PVR |
| Serotonin | Platelets/endothelium | PASMC proliferation + vasoconstriction | Remodeling |
| PDGF, VEGF | Endothelium | Endothelial/VSMC proliferation | Remodeling |
| Ca²⁺ (cytosolic ↑) | PASMCs | Vasoconstriction + proliferation | Sustained ↑ PVR |
Key Histopathological Findings (Morphology)
All forms of PH share:
- Medial hypertrophy of pulmonary muscular and elastic arteries
- Right ventricular hypertrophy
PAH-specific:
- Plexiform lesions (characteristic but not pathognomonic)
- Concentric laminar intimal proliferation
- In situ thrombosis
- Extension of muscle into normally non-muscular vessels
CTEPH:
- Organizing or recanalized thrombi in proximal and segmental pulmonary arteries
(Robbins & Cotran Pathologic Basis of Disease)
Hemodynamic Definition Recap
PH = mPAP ≥ 25 mmHg at rest (by right heart catheterization)
- Precapillary PH: mPAP ≥ 25, PCWP ≤ 15 mmHg, PVR > 3 Wood units (Groups 1, 3, 4, 5)
- Postcapillary PH: mPAP ≥ 25, PCWP > 15 mmHg (Group 2 - left heart disease)
- Combined pre- and postcapillary PH: mPAP ≥ 25, PCWP > 15, diastolic pressure gradient (DPG) ≥ 7 or PVR > 3
Sources:
- Fishman's Pulmonary Diseases and Disorders - Chapter 72: Pulmonary Vascular Structural and Functional Changes in PH; Pathogenic Mechanisms; Genetic, Cellular, and Molecular Mechanisms of PAH
- Robbins & Cotran Pathologic Basis of Disease - Chapter 15: Pathogenesis of Pulmonary Hypertension
- Fuster and Hurst's The Heart, 15th ed. - Chapter 57: Pathways in the Pathophysiology of PAH
- Recent review: Guignabert C et al. "Pathology and pathobiology of pulmonary hypertension: current insights and future directions." Eur Respir J. 2024 Oct. [PMID: 39209474]
This is a shared conversation. Sign in to Orris to start your own chat.