Explain Parkinson's disease
Reading File
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND ("pathophysiology" OR "treatment")
Searching the Web
Parkinson's disease pathophysiology mechanisms 2024 2025
Reading File
Reading File
Searching Images
Parkinson's disease dopaminergic pathway substantia nigra basal ganglia
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
I now have comprehensive content from Harrison's, Robbins, Adams & Victor's, and Bradley & Daroff's across pathophysiology, clinical features, genetics, and treatment. I have enough to write a thorough, well-cited answer.
Parkinson's Disease
Overview
Parkinson's disease (PD) is the second most common age-related neurodegenerative disorder, after Alzheimer's disease. Globally, approximately 10.8 million people are affected, a number expected to double within 20 years as the population ages. Mean age of onset is around 60 years; lifetime risk is ~3% in men and ~2% in women. James Parkinson first described the condition in 1817, famously characterizing it after observing just a handful of patients — some merely on the street. — Harrison's Principles of Internal Medicine 22E
Pathology
The hallmark pathological findings are:
- Loss of pigmented dopaminergic neurons in the substantia nigra pars compacta (SNc), with visible pallor of the substantia nigra and locus coeruleus on gross examination.
- Lewy bodies — round, eosinophilic cytoplasmic inclusions in surviving neurons, composed of filaments of α-synuclein, neurofilaments, and ubiquitin.
- Lewy neurites — dystrophic neuronal processes also containing aggregated α-synuclein.
The dopaminergic neurons of the SNc project to the striatum (caudate + putamen) via the nigrostriatal pathway, and their loss depletes striatal dopamine, disrupting the basal ganglia motor circuit. — Robbins & Kumar Basic Pathology
Neurodegeneration is not confined to dopaminergic neurons. Lewy pathology also affects:
- Cholinergic neurons of the nucleus basalis of Meynert
- Norepinephrine neurons of the locus coeruleus
- Serotonin neurons in the raphe nuclei
- Peripheral autonomic nervous system and enteric nervous system
This nondopaminergic pathology accounts for the many nonmotor features of PD. — Harrison's Principles of Internal Medicine 22E
Basal Ganglia Circuit Disruption
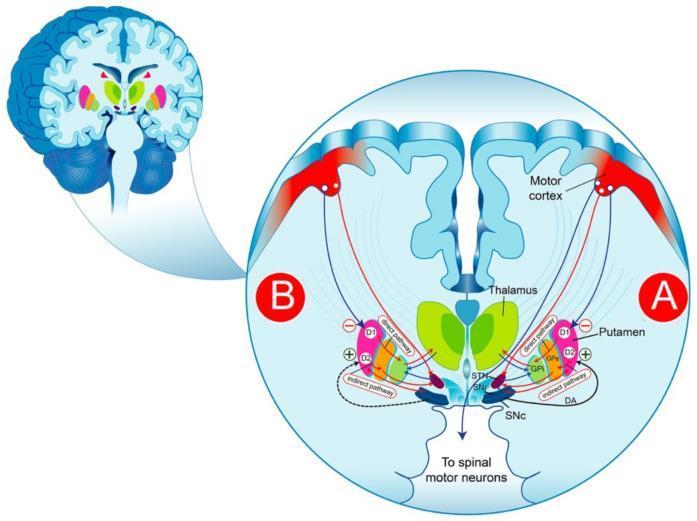
In the normal state, dopamine from the SNc acts on striatal neurons:
- Via D1 receptors → activates the direct pathway (striatum → GPi/SNr) → facilitates movement
- Via D2 receptors → inhibits the indirect pathway (striatum → GPe → STN → GPi) → reduces inhibition of movement
In PD, dopamine depletion tips the balance:
- The direct pathway is underactive
- The indirect pathway is overactive → increased STN activity → excessive GPi inhibition of the thalamus → reduced thalamocortical drive → bradykinesia and motor suppression
The subthalamic nucleus (STN) becomes the primary surgical target for deep brain stimulation (DBS) precisely because of this overactivity. — Bradley and Daroff's Neurology in Clinical Practice
Pathogenesis
Molecular mechanisms
| Mechanism | Detail |
|---|---|
| α-synuclein aggregation | α-Synuclein, a normal synaptic protein, misfolds and aggregates into Lewy body filaments; in high concentrations, soluble α-synuclein converts to toxic fibrils |
| Mitochondrial dysfunction | Impaired oxidative phosphorylation contributes to neuronal death (the neurotoxin MPTP reproduces parkinsonism by poisoning complex I of the mitochondrial respiratory chain) |
| Defective protein clearance | Abnormal autophagy and lysosomal degradation allow aggregates to accumulate; several PD-linked genes (Parkin, PINK1) function in mitophagy and endosomal trafficking |
| Neuroinflammation | Microglial activation and neuroinflammation amplify dopaminergic neuron loss |
Genetics
Most cases (~90%) are sporadic, but ~10–15% have a genetic basis:
| Gene | Inheritance | Protein/Function |
|---|---|---|
| SNCA (α-synuclein) | Autosomal dominant | Point mutations or gene duplications/triplications |
| LRRK2 | Autosomal dominant | Most common cause of familial PD; kinase function unclear |
| PARKIN | Autosomal recessive | E3 ubiquitin ligase; mitophagy |
| PINK1 | Autosomal recessive | Mitochondrial serine/threonine kinase |
| DJ-1 | Autosomal recessive | Oxidative stress sensor |
| GBA1 | Risk factor | Glucocerebrosidase; most common genetic risk factor for sporadic PD |
— Robbins & Kumar Basic Pathology; Adams and Victor's Principles of Neurology, 12th Ed.
Braak Staging Hypothesis
Braak and colleagues proposed that PD pathology begins outside the substantia nigra — earliest changes appear in the enteric nervous system, dorsal motor nucleus of the vagus, and olfactory bulb, then spread rostrally to the brainstem and eventually the SNc (stage 3–4) and neocortex (stages 5–6). This "gut-first" hypothesis is supported by epidemiological data showing that truncal vagotomy reduces PD incidence. — Adams and Victor's Principles of Neurology; Harrison's
Clinical Features
Cardinal motor features ("TRAP")
| Feature | Description |
|---|---|
| Tremor | Resting tremor (4–6 Hz), "pill-rolling"; suppressed by voluntary movement |
| Rigidity | Lead-pipe or cogwheel resistance throughout range of motion |
| Akinesia/Bradykinesia | Slowness and poverty of movement; micrographia, hypomimia, hypophonia |
| Postural instability | Late feature; predisposes to falls |
Other motor features
- Freezing of gait (sudden inability to initiate steps)
- Festination (progressively quickening, shuffling gait)
- Reduced arm swing
- Masked facies, reduced blinking, drooling, dysphagia
Nonmotor features
These frequently predate motor symptoms by years and constitute the prodromal phase:
| Domain | Examples |
|---|---|
| Autonomic | Orthostatic hypotension, constipation, urinary dysfunction, sexual dysfunction, seborrhea |
| Sensory | Hyposmia/anosmia (often earliest sign), pain |
| Psychiatric | Depression (most common; may predate motor onset), anxiety, apathy, psychosis (visual hallucinations, usually drug-related) |
| Sleep | REM sleep behavior disorder (RBD), insomnia, excessive daytime somnolence |
| Cognitive | PD-MCI → Parkinson's disease dementia (PDD) in the majority over time |
— Harrison's Principles of Internal Medicine 22E; Bradley and Daroff's Neurology in Clinical Practice
Diagnosis
PD is a clinical diagnosis. No single test is definitive. Features supporting diagnosis:
- Asymmetric onset
- Rest tremor
- Clear response to dopaminergic therapy
Imaging:
- DaT-SPECT (dopamine transporter scan): shows reduced striatal dopamine transporter binding; useful to distinguish PD from essential tremor, but cannot distinguish PD from other parkinsonian syndromes
- FDG-PET and structural MRI are used mainly to exclude other conditions
Differential Diagnosis (Parkinson-Plus Syndromes)
| Condition | Distinguishing Features |
|---|---|
| Progressive supranuclear palsy (PSP) | Vertical gaze palsy (especially downward), axial rigidity, early falls, "hummingbird sign" on MRI |
| Multiple system atrophy (MSA) | Prominent autonomic failure, cerebellar signs, poor levodopa response |
| Corticobasal syndrome (CBS) | Asymmetric dystonia, alien limb phenomenon, cortical sensory loss |
| Drug-induced parkinsonism | Symmetric, history of dopamine antagonists (neuroleptics, metoclopramide) |
| Dementia with Lewy bodies (DLB) | Dementia precedes or co-emerges with parkinsonism; prominent hallucinations, fluctuating cognition |
Treatment
Pharmacotherapy
Levodopa + carbidopa remains the gold standard — most effective symptomatic treatment. Carbidopa inhibits peripheral decarboxylation, allowing more levodopa to cross the blood-brain barrier. Long-term complications include motor fluctuations ("wearing off") and dyskinesias.
| Drug Class | Examples | Notes |
|---|---|---|
| Levodopa formulations | Sinemet, Rytary (extended-release), Duopa (intestinal gel) | Most potent; dyskinesia risk with long-term use |
| Dopamine agonists | Pramipexole, ropinirole, rotigotine patch | Used alone early or as adjunct; side effects: impulse control disorders, somnolence |
| MAO-B inhibitors | Selegiline, rasagiline, safinamide | Reduce dopamine breakdown; mild symptomatic benefit; possible neuroprotective effect debated |
| COMT inhibitors | Entacapone, opicapone | Extend levodopa effect; used for wearing-off |
| Anticholinergics | Trihexyphenidyl | Mainly for tremor in younger patients; cognitive side effects |
| Amantadine | — | Antidyskinesia; also mild symptomatic benefit |
Surgical / Neuromodulation
- Deep brain stimulation (DBS) of the subthalamic nucleus (STN) or globus pallidus interna (GPi): highly effective for motor fluctuations and dyskinesias in patients who have a good levodopa response; does not modify disease progression.
- Focused ultrasound thalamotomy: non-invasive ablation, primarily for tremor.
- rTMS: investigational; modest and inconsistent motor benefit reported. — Bradley and Daroff's Neurology
Neuroprotective strategies
No therapy has yet been proven to slow or halt progression in humans. Trials of nilotinib, GLP-1 agonists, and LRRK2 inhibitors are ongoing.
Non-pharmacological
- Exercise (high-intensity aerobic and resistance training) has the most robust evidence for slowing functional decline
- Speech therapy (Lee Silverman Voice Treatment)
- Physiotherapy, occupational therapy
- Deep diaphragmatic breathing and aspiration precautions for dysphagia
Prognosis
PD is a progressive disease with no cure. Motor symptoms tend to worsen over years; nonmotor features (especially dementia and autonomic failure) become dominant in late stages and are major drivers of disability and nursing home placement. Median survival from diagnosis is approximately 10–15 years, though this varies widely.
Sources: Harrison's Principles of Internal Medicine 22E (2025); Robbins & Kumar Basic Pathology; Adams and Victor's Principles of Neurology, 12th Ed.; Bradley and Daroff's Neurology in Clinical Practice
This is a shared conversation. Sign in to Orris to start your own chat.