Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
I now have all the material needed for a comprehensive, well-illustrated answer. Here is a structured explanation of the mechanism of ARDS.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition & Berlin Criteria
ARDS is a clinical syndrome of acute respiratory insufficiency caused by diffuse alveolar damage (DAD). The Berlin definition requires:
- Bilateral pulmonary infiltrates on CXR/CT
- PaO₂/FiO₂ (P/F) ratio <300 mmHg (mild), <200 (moderate), <100 (severe)
- Known clinical insult within 7 days
- Not fully explained by cardiac failure or fluid overload
— Sabiston Textbook of Surgery, p. 940
Causes (Triggers)
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia (bacterial, viral) | Sepsis (most common overall) |
| Aspiration | Severe trauma / shock |
| Inhalation injury | Pancreatitis |
| Near-drowning | Massive transfusion (TRALI) |
— Robbins & Kumar Basic Pathology, p. 1735; Harrison's, p. 2343
Phases of ARDS

Time course of ARDS: exudative → proliferative → fibrotic — Harrison's Principles of Internal Medicine, 22e
Phase 1: Exudative Phase (Days 0–7)
This is the core pathophysiological phase. The key events are:
1. Initial Insult → Pattern Recognition & Innate Immune Activation
- Pathogens, DAMPs (damage-associated molecular patterns), or toxins are recognized by Toll-like receptors (TLRs) on alveolar type I (ATI) epithelial cells and resident alveolar macrophages.
- Macrophages secrete pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-8/CXCL8), initiating a cytokine cascade.
2. Neutrophil Recruitment & Activation (Central Mediators)
Neutrophils have a central role in endothelial and epithelial injury:
- IL-8 and other chemokines recruit circulating PMNs from the bloodstream into the alveolar interstitium and airspaces.
- Activated neutrophils migrate across the endothelium and epithelium, releasing:
- Proteases (elastase, matrix metalloproteinases) — digest basement membrane and tight junction proteins
- Reactive oxygen species (ROS) — oxidant-mediated membrane and organelle injury
- Neutrophil extracellular traps (NETs) — chromatin-protease complexes that amplify injury
3. Endothelial & Epithelial Barrier Disruption
The alveolar-capillary membrane consists of two barriers:
| Barrier | Injury Consequence |
|---|---|
| Pulmonary capillary endothelium | Increased vascular permeability → protein-rich fluid leaks into interstitium |
| Alveolar epithelium (type I cells) | Necrosis/apoptosis → fluid floods the airspace |
- Type I pneumocytes (gas exchange cells, ~95% of alveolar surface area) are highly vulnerable to injury and lack regenerative capacity.
- Disruption of tight junctions and ENaC (epithelial sodium channels) impairs active fluid clearance from alveoli, compounding edema.
4. Protein-Rich Alveolar Edema
- Plasma proteins (including fibrinogen) flood the airspace.
- Fibrinogen is converted to fibrin, which precipitates on the denuded alveolar walls along with necrotic cell debris → forming hyaline membranes (the histologic hallmark of DAD).
- Surfactant dysfunction: Plasma proteins inactivate surfactant; type II pneumocyte injury reduces surfactant production. Loss of surfactant increases alveolar surface tension → alveolar collapse and atelectasis.
5. Additional Injury Pathways
- Monocytes migrate into the lung and cause epithelial apoptosis via IFN-β–dependent TRAIL (TNF-related apoptosis-inducing ligand) release.
- Platelets form aggregates with PMNs (PMN-platelet aggregates), amplifying NET formation and oxidant injury.
- Red blood cells that extravasate release cell-free hemoglobin, causing further oxidant-mediated damage.
- Microvascular thrombi form due to local coagulation activation, contributing to increased pulmonary dead space and hypercapnia.
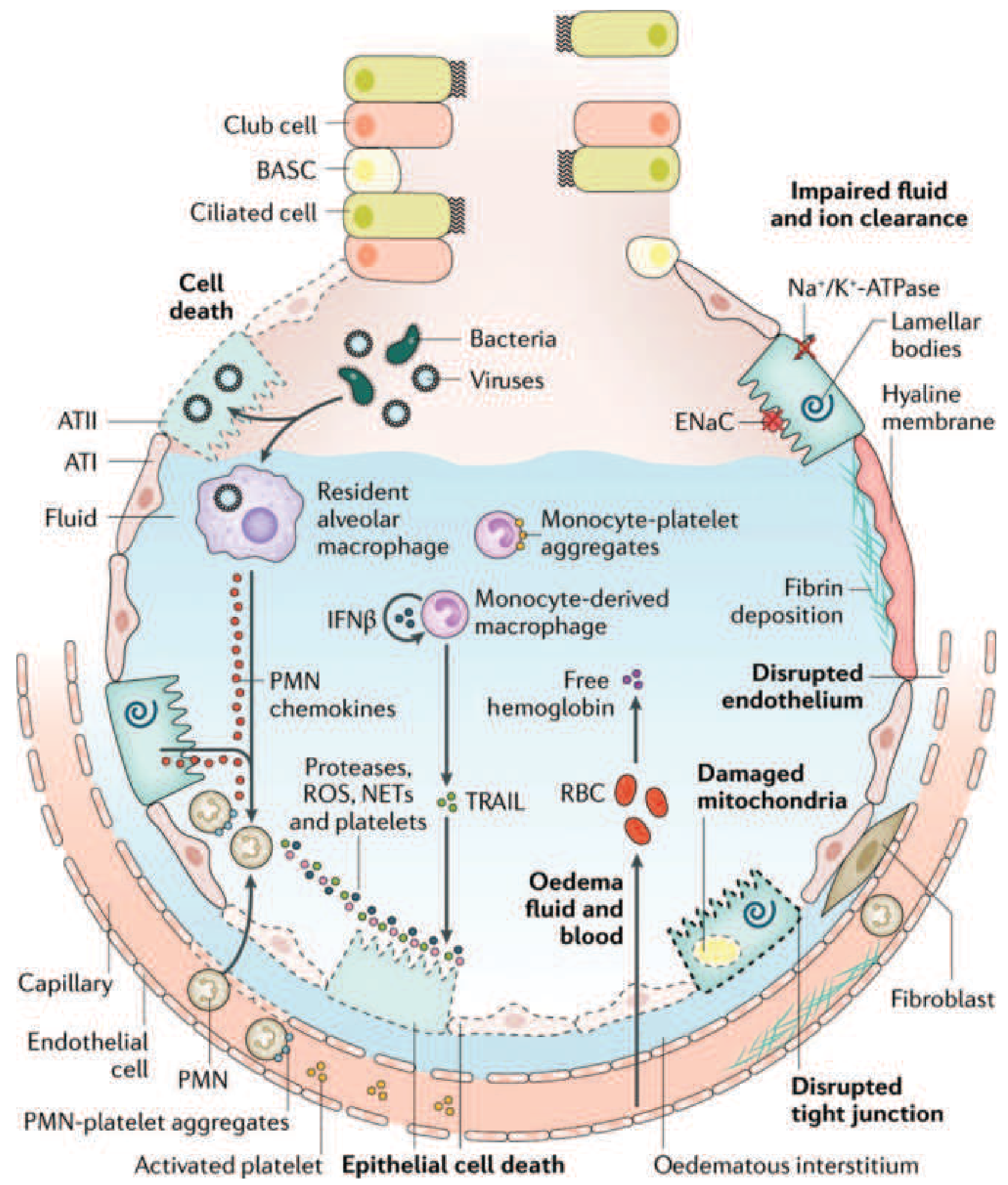
Figure 312-3 from Harrison's 22e: Injured alveolus in the acute phase of ARDS. PMNs release proteases, ROS, and NETs. Monocytes cause epithelial apoptosis via TRAIL. Platelet and RBC extravasation amplify injury. Disrupted tight junctions, impaired ENaC, and damaged Na⁺/K⁺-ATPase impair edema clearance.
Physiologic Consequences of the Exudative Phase
- Hypoxemia: Edema-filled alveoli cannot participate in gas exchange → intrapulmonary shunting (perfusion without ventilation); refractory to supplemental O₂ alone.
- Decreased compliance: Fluid and collapse stiffen the lung ("baby lung" concept — only a small proportion of alveoli remain aerated and recruitable).
- Increased dead space: Microvascular obstruction impairs CO₂ excretion → hypercapnia.
- Bilateral infiltrates on CXR/CT, predominantly in dependent zones due to gravitational distribution of edema.
— Harrison's Principles of Internal Medicine 22e, pp. 2343–2344; Goldman-Cecil Medicine, p. 4317
Phase 2: Proliferative Phase (Days 7–21)
- Neutrophil infiltrate is replaced by lymphocyte-predominant inflammation.
- Type II pneumocytes proliferate along denuded basement membranes — they synthesize new surfactant and differentiate into type I cells to restore the epithelial barrier.
- Alveolar exudates begin to organize; macrophages clear fibrin and debris.
- Many patients improve during this phase.
— Harrison's Principles of Internal Medicine 22e, p. 2344
Phase 3: Fibrotic Phase (>Day 21, subset of patients)
- Alveolar edema and inflammatory exudates convert to alveolar-duct and interstitial fibrosis.
- Disrupted acinar architecture leads to emphysema-like changes and large bullae.
- Intimal fibroproliferation in pulmonary microvessels → progressive vascular occlusion → pulmonary hypertension.
- Results in reduced compliance, increased dead space, elevated pneumothorax risk, and oxygen dependence.
— Harrison's Principles of Internal Medicine 22e, p. 2344
Summary Cascade
Trigger (sepsis, trauma, aspiration...)
↓
TLR activation on ATI cells & macrophages
↓
Cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
PMN recruitment & activation into alveoli
↓
Proteases + ROS + NETs released
↓
Endothelial & epithelial barrier disruption
├── Protein-rich edema floods alveoli
├── Fibrin deposition → hyaline membranes
├── Surfactant inactivation → alveolar collapse
└── Impaired Na/fluid clearance (ENaC/ATPase)
↓
Hypoxemia (shunt) + Reduced compliance + Dead space
↓
Respiratory failure (ARDS)
Key Points
| Feature | Detail |
|---|---|
| Hallmark histology | Diffuse alveolar damage (DAD) with hyaline membranes |
| Central cellular mediator | Neutrophils (PMNs) |
| Key cytokines | TNF-α, IL-1β, IL-6, IL-8, IFN-β |
| Edema type | Non-cardiogenic (high-protein, permeability edema) |
| Surfactant | Inactivated by plasma proteins + reduced production |
| Gas exchange defect | Intrapulmonary shunt → refractory hypoxemia |
| Phases | Exudative → Proliferative → Fibrotic |
Sources:
- Harrison's Principles of Internal Medicine, 22e, Chapter 312
- Robbins & Kumar Basic Pathology, Chapter 11
- Goldman-Cecil Medicine, Chapter 79 / Chapter 91
- Sabiston Textbook of Surgery, Chapter 11
This is a shared conversation. Sign in to Orris to start your own chat.