Inflammation
Reading File
Finding Sources
Searching PubMed
"inflammation" AND "mechanisms"
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have sufficient content to write a comprehensive, well-structured answer on inflammation from Robbins & Kumar Basic Pathology. Let me compile the full response.
Inflammation
Inflammation is the response of vascularized tissues to infection, cell injury, or necrosis. Its purpose is to eliminate the initial cause of injury, clear out dead cells and tissues, and initiate tissue repair. The cardinal signs are redness (rubor), heat (calor), swelling (tumor), pain (dolor), and loss of function - described by Celsus and Virchow.
Inflammation is broadly divided into acute and chronic forms.
Acute Inflammation
Acute inflammation is rapid in onset (seconds to minutes), of short duration (hours to days), and characterized by:
- Dilation of small vessels - increasing blood flow
- Increased vascular permeability - enabling plasma proteins and leukocytes to exit the circulation
- Emigration of leukocytes (mainly neutrophils) to the site of injury
Vascular Reactions
Vasodilation is one of the earliest events, mediated chiefly by histamine acting on vascular smooth muscle. It involves mainly postcapillary venules and is responsible for the heat and redness at the inflamed site.
Increased permeability follows quickly, driven by contraction of endothelial cells (creating interendothelial gaps). This is elicited by histamine, bradykinin, and leukotrienes. It occurs within 15-30 minutes of exposure. The result is an exudate - protein-rich fluid that leaks into the tissue.
Exudate vs. Transudate
- Exudate: High protein, may contain cellular debris and WBCs. Implies increased vascular permeability (inflammatory).
- Transudate: Low protein (mostly albumin), few cells. Due to osmotic/hydrostatic imbalance - NOT inflammation (e.g., heart failure, cirrhosis, nephrotic syndrome).
- Pus (purulent exudate): Rich in neutrophils, dead cell debris, and microbes.
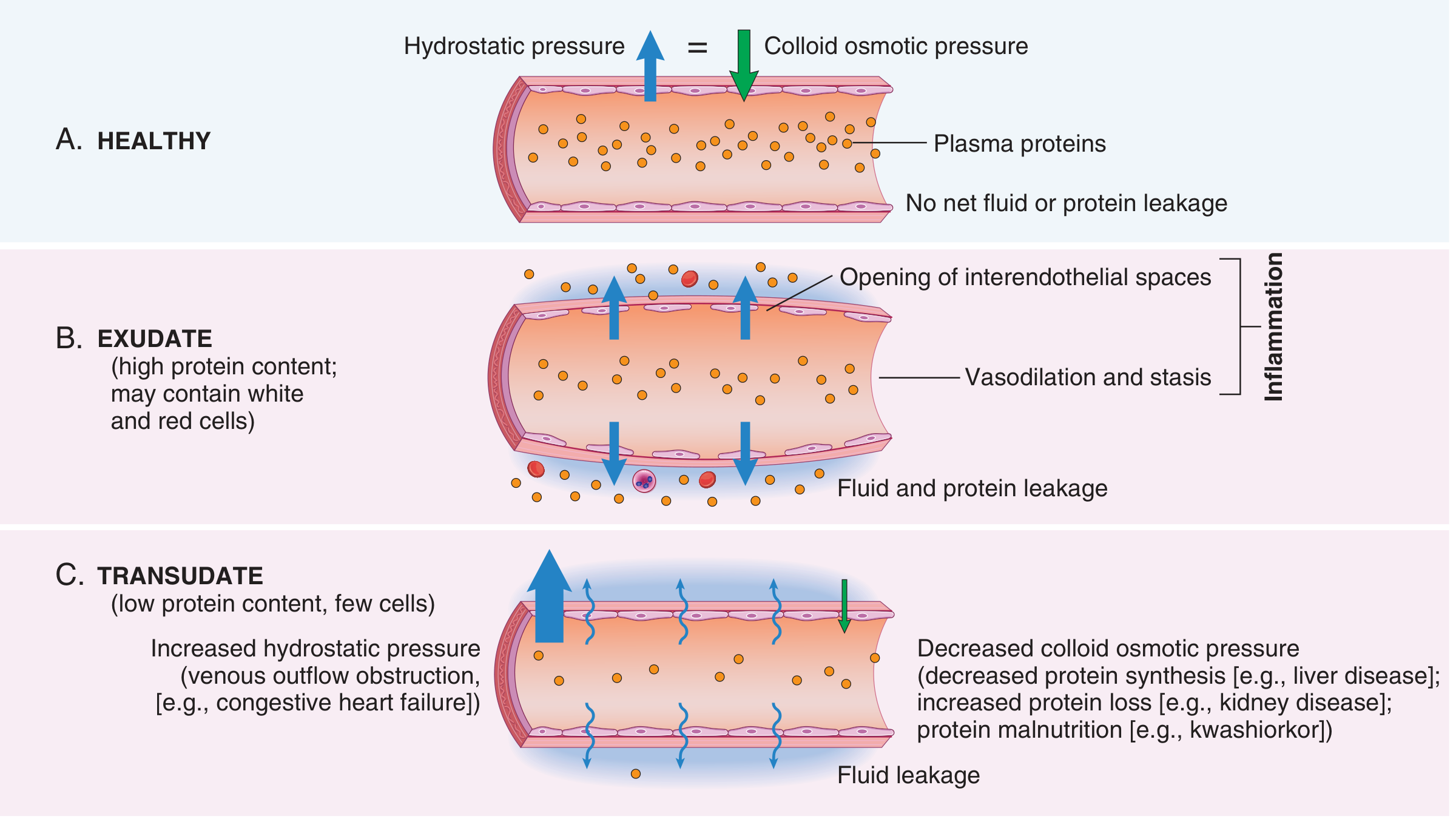
The loss of fluid plus vasodilation slows blood flow (stasis), concentrates red cells, and raises blood viscosity - seen grossly as vascular congestion.
Cellular Events: Leukocyte Recruitment
Leukocyte emigration proceeds in a sequence:
| Step | What Happens |
|---|---|
| Margination | Neutrophils move to the vessel periphery as blood slows |
| Rolling | Transient adhesion mediated by selectins (E-selectin, P-selectin on endothelium; L-selectin on leukocytes) |
| Firm adhesion | Integrins on leukocytes bind to ICAMs on endothelium, activated by chemokines like IL-8 |
| Transmigration (diapedesis) | Leukocytes squeeze through interendothelial junctions (mainly via PECAM-1/CD31) |
| Chemotaxis | Migration toward chemoattractants: bacterial products (fMLP), complement C5a, leukotriene B4, IL-8 |
Once at the site, leukocytes (primarily neutrophils) perform phagocytosis - enhanced by opsonization with IgG and C3b. Inside the phagolysosome, killing occurs via:
- Reactive oxygen species (ROS) - NADPH oxidase generates superoxide → H₂O₂ → hypochlorous acid (via myeloperoxidase)
- Nitric oxide (NO)
- Granule enzymes - lysozyme, defensins, proteases, elastase, cathepsin G
Neutrophils also form NETs (Neutrophil Extracellular Traps) - chromatin-histone networks that trap and kill microbes extracellularly, at the cost of the neutrophil's life.
Chemical Mediators of Inflammation
Mediators are produced or activated at the injury site and control the inflammatory response:
Cell-derived mediators:
| Mediator | Source | Key Actions |
|---|---|---|
| Histamine | Mast cells, basophils, platelets | Vasodilation, increased permeability (earliest) |
| Prostaglandins (PGE2, PGI2) | Arachidonic acid via COX | Vasodilation, pain, fever |
| Leukotrienes (LTB4, LTC4, LTD4) | Arachidonic acid via lipoxygenase | Chemotaxis (LTB4), bronchoconstriction, permeability (LTC4/D4) |
| Platelet-activating factor (PAF) | Leukocytes, endothelium | Platelet aggregation, vasodilation, permeability |
| TNF, IL-1 | Macrophages | Fever, acute-phase response, leukocyte adhesion, systemic effects |
| IL-8 (CXCL8) | Macrophages, endothelium | Powerful neutrophil chemokine |
| Nitric oxide | Endothelium, macrophages | Vasodilation, microbial killing |
Plasma-derived mediators:
| System | Key Products | Actions |
|---|---|---|
| Complement | C3a, C5a (anaphylatoxins); C5b-9 (MAC) | Vasodilation, chemotaxis, phagocytosis, cell lysis |
| Kinin system | Bradykinin | Vasodilation, permeability, pain |
| Coagulation | Thrombin, fibrin | Platelet activation, permeability |
Key arachidonic acid pathway targets are exploited pharmacologically:
- NSAIDs (aspirin, ibuprofen) block COX → reduce prostaglandins
- Corticosteroids block phospholipase A2 (prevent arachidonic acid release) and suppress many mediator genes
- Montelukast blocks leukotriene receptors
Outcomes of Acute Inflammation
Three possible outcomes:
- Complete resolution - usual outcome if injury is brief, tissue is capable of regeneration. Involves macrophage clearance of debris and lymphatic resorption of edema.
- Healing by fibrosis (scarring) - when tissue destruction is extensive, tissue cannot regenerate, or fibrin exudate cannot be cleared.
- Progression to chronic inflammation - when the injurious agent persists or healing is impaired.
Chronic Inflammation
Chronic inflammation is a prolonged (weeks to months) response in which inflammation, tissue injury, and repair attempts coexist simultaneously. It may follow unresolved acute inflammation or begin insidiously without an acute phase.
Causes
- Persistent infections: Mycobacteria, fungi, parasites, viruses - organisms that are hard to eradicate
- Hypersensitivity/autoimmune diseases: Rheumatoid arthritis, multiple sclerosis, asthma - self-sustaining immune activation
- Prolonged toxic exposure: Silica inhalation (silicosis), excess cholesterol/lipids (atherosclerosis)
Morphologic Features (vs. Acute)
| Feature | Acute | Chronic |
|---|---|---|
| Duration | Hours to days | Weeks to months |
| Dominant cell | Neutrophils | Macrophages, lymphocytes, plasma cells |
| Vascular changes | Prominent | Less prominent |
| Tissue damage + repair | Repair follows | Both occur simultaneously |
| Fibrosis | Absent initially | Often present |
Key Cells in Chronic Inflammation
Macrophages are the dominant cells. They:
- Destroy foreign material and tissue debris via phagocytosis
- Secrete cytokines (TNF, IL-1, IL-12), growth factors (PDGF, FGF), and enzymes
- Activate T cells and are activated by them in return (classical M1 activation)
- Can also be alternatively activated (M2) to promote wound healing and fibrosis via IL-4 and IL-13
Lymphocytes: CD4+ T cells (Th1, Th2, Th17) activate macrophages and coordinate adaptive immunity. CD8+ T cells kill infected cells. B cells produce antibodies.
Plasma cells: Produce antibodies against persistent antigens.
Eosinophils: Prominent in parasitic infections and allergic conditions.
Mast cells: Found in connective tissues; release histamine and cytokines.
Granulomatous Inflammation
A distinctive pattern of chronic inflammation characterized by aggregates of activated macrophages called epithelioid cells, often with:
- Multinucleated giant cells (fusion of macrophages) - Langhans type (nuclei at periphery) or foreign-body type
- Peripheral rim of lymphocytes and fibroblasts
- Central caseous necrosis (in tuberculosis)
Causes of granulomas ("BFS CATS"):
- Berylliosis
- Foreign bodies (sutures, talc)
- Sarcoidosis (non-caseating)
- Crohn's disease
- Aspergillus / fungal infections
- Tuberculosis (caseating)
- Syphilis
Granuloma formation requires sustained macrophage activation by T-cell-derived IFN-γ. The goal is to "wall off" what cannot be destroyed.
Morphological Patterns of Acute Inflammation
| Pattern | Description | Example |
|---|---|---|
| Serous | Watery fluid, low cell content | Blister, pleural effusion |
| Fibrinous | Fibrin-rich exudate | Pericarditis ("bread and butter"), lobar pneumonia |
| Purulent/suppurative | Pus (neutrophils + debris) | Abscess, furuncle |
| Ulcerative | Necrosis + sloughing of inflamed surface | Peptic ulcer, aphthous stomatitis |
| Pseudomembranous | Membrane of fibrin/necrotic debris on mucosal surface | C. diff colitis, diphtheria |
Systemic Effects of Inflammation (Acute Phase Response)
Cytokines (especially IL-1, TNF, IL-6) drive systemic changes:
- Fever: Cytokines act as pyrogens, triggering prostaglandin (PGE2) synthesis in the hypothalamus
- Leukocytosis: Bone marrow is stimulated to release more WBCs; bacterial infections favor neutrophilia, viral infections favor lymphocytosis, parasites/allergies favor eosinophilia
- Acute-phase proteins (liver-derived): C-reactive protein (CRP), serum amyloid A, fibrinogen, hepcidin - elevated in inflammation
- Elevated ESR: Due to increased fibrinogen causing RBC rouleaux formation
- Anorexia, fatigue, somnolence: Mediated by cytokines on the CNS
- Septic shock (in severe infection): Vasodilation, DIC, multi-organ failure - driven by massive TNF/IL-1 release
Tissue Repair
Following resolution of inflammation, tissue is repaired by:
- Regeneration - replacing damaged cells with cells of the same type (e.g., hepatocytes in the liver)
- Scarring (fibrosis) - connective tissue fills the defect when tissue cannot regenerate
Key mediators of repair:
- VEGF (produced by macrophages under hypoxic conditions via HIF-1α) drives angiogenesis - sprouting of new capillaries into the repair zone
- TGF-β is the main driver of fibroblast activation and collagen deposition
- PDGF stimulates fibroblast migration and proliferation
- Granulation tissue = proliferating fibroblasts + new capillaries + ECM - forms the scaffold of repair
Wound healing:
- First intention (primary union): Clean, apposed wound edges; minimal scarring
- Second intention (secondary union): Open wounds, extensive granulation tissue, more scarring and wound contraction (myofibroblasts)
Key Pharmacological Targets
| Drug Class | Target | Effect |
|---|---|---|
| NSAIDs | COX-1/COX-2 | Block prostaglandin synthesis; reduce pain, fever, inflammation |
| Aspirin | COX irreversibly | Also inhibits platelet TXA2 |
| Glucocorticoids | PLA2, NF-κB, cytokine genes | Broad anti-inflammatory |
| Anti-TNF biologics | TNF-α | Used in RA, IBD, psoriasis |
| Anti-IL-1 (Anakinra) | IL-1 receptor | Autoinflammatory diseases, RA |
| Canakinumab | IL-1β | Gout, CAPS, recently studied in CV disease |
| Colchicine | Tubulin polymerization | Blocks neutrophil migration; gout, pericarditis |
Source: Robbins & Kumar Basic Pathology, Chapters 2-3
Source: Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 3
This is a shared conversation. Sign in to Orris to start your own chat.