I want to make a note on, you know, the T cell maturation, where how it happens in the thymus with double selection, negative selection, and everything. You know, the positive, first, the developed T cell, which is an immature T lymphocyte, enters into the thymus first, into the cortical region, and it is the double positive cell. When it comes, it has both, you know, CD, TBC, it has a CD8 positive receptor, CD4 positive receptor, and a Treg receptor. So saying it is double positive, you know, from both. So for it to become from double positive to single positive, it has to go through a cortical positive selection, where we check whether are you binding, are you able to bind to the self-antigen of MHC and recognizing the MHC and the antigen. Yes, if you are recognizing it, then it will go through the next process of single positivity, where the cell is either CD4 or CD8. And after that, through the single positivity, it will have to go outside. Before going that outside the thymus, it will undergo negative selection, where it will ask whether, how strong and how avidly are you binding to the self-antigen. So these self-antigens are actually exposed to lymphocytes through the dendritic cells within the medulla of the thymus, presenting the self-antigens and seeing whether they are binding it. And another cell is the medullary epithelial cell. Inside that medullary epithelial cell, there is a mechanism of a nuclear receptor called IR, through which there are some self-antigens expressed on the medullary epithelial cell. So when there is an IR mutation, where some self-antigens are not expressed on the medullary epithelial cell, so the cell escapes. The same lymphocytes have never exposed to this self-antigen. So when they escape, they can become, they can cause this, this is called autoimmune polyglandular syndrome, where this T-cells will attack autoimmunely the endocrine glands. So this concept I want. And correct me if I'm not and make a USMLE topper level note.
T cell maturation thymus positive selection negative selection double positive single positive CD4 CD8
AIRE gene medullary thymic epithelial cells negative selection autoimmune polyglandular syndrome
T cell maturation thymus positive negative selection diagram
T Cell Maturation in the Thymus
Overview
Stage 0: The Arriving Cell — Double Negative (DN)
Correction on your note: You mentioned the cell arrives as double positive — technically it arrives as double negative and becomes double positive within the cortex after TCR rearrangement. The DP stage is where positive selection then acts on the cell.
Stage 1: Positive Selection (Cortex)
| Outcome | Fate |
|---|---|
| TCR binds self-MHC with appropriate (moderate) affinity | ✅ Survives → proceeds to single positive stage |
| TCR cannot bind self-MHC at all | ❌ Dies by neglect (apoptosis — no survival signal) |
- If TCR recognizes MHC Class I → downregulates CD4 → becomes CD8+ single positive
- If TCR recognizes MHC Class II → downregulates CD8 → becomes CD4+ single positive
Stage 2: Negative Selection (Medulla)
- Medullary dendritic cells
- Medullary thymic epithelial cells (mTECs)
| Outcome | Fate |
|---|---|
| TCR binds self-antigen with high avidity | ❌ Clonal deletion (apoptosis) — eliminates autoreactive T cells |
| TCR binds self-antigen with low/moderate avidity | ✅ Survives → exits thymus as a naive, self-tolerant T cell |
The AIRE Gene — The Key to Negative Selection in the Medulla
What is AIRE?
Correction on your note: You referred to it as "IR" — the correct name is AIRE (Autoimmune Regulator gene), located on chromosome 21.
Function of AIRE
AIRE Mutation → Autoimmune Polyglandular Syndrome Type 1 (APS-1)
Mechanism
AIRE mutation (loss of function)
↓
mTECs fail to express tissue-specific self-antigens
↓
Autoreactive T cells are NOT deleted during negative selection
↓
Autoreactive T cells escape to periphery
↓
Attack endocrine (and other) glands → APS-1
Genetics
| Feature | Detail |
|---|---|
| Inheritance | Autosomal recessive (classic form) |
| Gene | AIRE gene, chromosome 21 |
| Some dominant mutations | Localized to the PHD1 domain |
| High-frequency ethnic groups | Finnish, Sardinians, Iranian Jews, Norwegians, Irish |
Classic APS-1 Clinical Triad (Whitaker's Triad)
| Component | Notes |
|---|---|
| Chronic mucocutaneous candidiasis | Usually first to appear, in childhood |
| Hypoparathyroidism | Most common endocrine manifestation |
| Primary adrenal insufficiency (Addison's disease) | Anti-21-hydroxylase antibodies |
Mnemonic: "The HAC" — Hypoparathyroidism, Adrenal insufficiency, Candidiasis
Full T Cell Maturation Flowchart
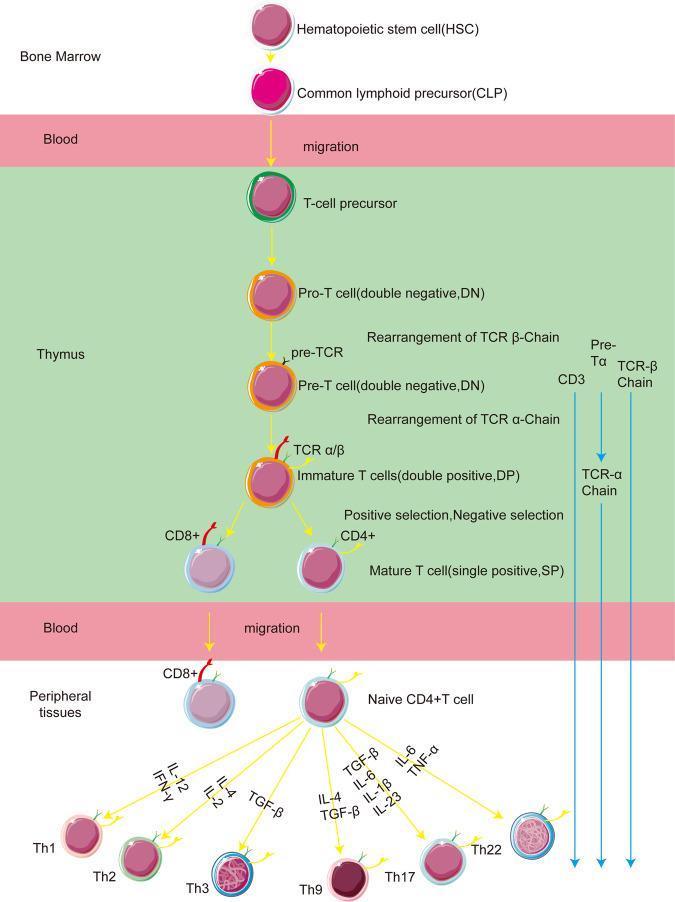
Bone Marrow
→ Blood
→ Thymus CORTEX
↓
T cell precursor
↓ (TCR rearrangement)
DOUBLE NEGATIVE (CD4⁻ CD8⁻)
↓ (upregulate both co-receptors)
DOUBLE POSITIVE (CD4⁺ CD8⁺)
↓
┌─────────────────────────────────────┐
│ POSITIVE SELECTION (cortex) │
│ cTECs test: can TCR bind self-MHC? │
│ Fail → die by neglect │
└─────────────────────────────────────┘
↓
SINGLE POSITIVE (CD4⁺ OR CD8⁺)
↓ migrates to medulla
┌──────────────────────────────────────────┐
│ NEGATIVE SELECTION (medulla) │
│ mTECs + DCs test: too autoreactive? │
│ AIRE → ectopic self-antigen expression │
│ High avidity → apoptosis (clonal delete) │
│ Low avidity → survives │
└──────────────────────────────────────────┘
↓
Naive, self-tolerant T cell → Periphery
High-Yield USMLE Summary
| Concept | Key Point |
|---|---|
| Double Positive cell | CD4⁺ CD8⁺ — in thymic cortex after TCR rearrangement |
| Positive selection | Cortex / cTECs / "Can you see self-MHC?" → survive or die by neglect |
| Negative selection | Medulla / mTECs + DCs / "Are you too self-reactive?" → clonal deletion |
| AIRE gene | Transcriptional regulator in mTECs; drives ectopic self-antigen expression |
| AIRE mutation | → Autoreactive T cells escape → APS-1 |
| APS-1 genetics | Autosomal recessive; chromosome 21 |
| APS-1 triad | Hypoparathyroidism + Adrenal insufficiency + Candidiasis |
Okay, now about it's very nice and the but remove the part of what I said wrong. I want, you know, just all together compressed notes. Remove whatever wrong I said, like, if you want to say flowcharts, please give a sentence-based arrow language flowcharts because I have to copy it in my notes. And no more floats, no more tables. I just wanted the bullet points with clear-cut concepts.
T Cell Maturation in the Thymus
Origin
- T cell precursors arise from hematopoietic stem cells in the bone marrow
- They migrate via blood to the thymus for maturation
Stage 1 — Double Negative (DN) Phase
- Arriving precursors are CD4⁻ CD8⁻ (double negative)
- Inside the thymic cortex, they undergo TCR gene rearrangement (β-chain first, then α-chain)
- After successful rearrangement, they upregulate both CD4 and CD8 → become Double Positive (DP)
Stage 2 — Positive Selection (Cortex)
- Occurs in the thymic cortex
- Mediated by cortical thymic epithelial cells (cTECs)
- Question asked: "Can your TCR bind self-MHC with at least moderate affinity?"
- Yes → cell survives and proceeds
- No → cell dies by neglect (apoptosis, no survival signal)
- DP cell now commits to a lineage:
- TCR recognizes MHC Class I → loses CD4 → becomes CD8⁺ single positive
- TCR recognizes MHC Class II → loses CD8 → becomes CD4⁺ single positive
- Single positive (SP) cell then migrates to the medulla
Stage 3 — Negative Selection (Medulla)
- Occurs in the thymic medulla
- Mediated by two cell types:
- Medullary dendritic cells
- Medullary thymic epithelial cells (mTECs)
- Question asked: "Are you binding self-antigens too strongly?"
- High avidity binding → clonal deletion (apoptosis) — eliminates autoreactive T cells
- Low/moderate avidity → cell survives → exits thymus as a mature, naive, self-tolerant T cell
AIRE Gene — The Engine of Negative Selection
- AIRE = Autoimmune Regulator gene, located on chromosome 21
- Expressed in mTECs
- AIRE is a transcriptional regulator that forces mTECs to ectopically express tissue-specific peripheral self-antigens (e.g., insulin, thyroglobulin) inside the thymus
- This exposes developing T cells to peripheral antigens they would otherwise never see
- If a T cell binds these antigens too strongly → deleted
- This process is called central tolerance
AIRE Mutation → APS-1
- Inheritance: Autosomal recessive (classic); some dominant mutations in the PHD1 domain
- Chromosome: 21
- Higher frequency in: Finns, Sardinians, Iranian Jews, Norwegians, Irish
APS-1 Clinical Triad
- Chronic mucocutaneous candidiasis — appears first, in childhood
- Hypoparathyroidism — most common endocrine feature
- Primary adrenal insufficiency (Addison's disease) — anti-21-hydroxylase antibodies
Full Maturation Flow
Things like this way now also tell me about the peripheral tolerance that happens peripherally somewhere because of anergy or apoptosis, you know, right? Like, let's say peripherally we have one T cell which has a T cell receptor on it and then it will go and attach to some dendritic, some antigen on the dendritic cell presenting MHC1, first signal is got activated, but for the second signal on the dendritic cell, the T cell should express CD28 which will bind to B7 receptor on the dendritic cell, then the second signal activates. But for some reason, because it's not activated, because it is not the self-reactive, it cannot recognize the self-antigen, has got a second reaction doesn't happen, then this cell shutdown becomes anergy, A-N-E-R-G-Y and cause cell death. If this does not happen, this is one way, if this does not happen, then the other way is like fast apoptosis pathway. The CD cell try to do it, but the second signal never happens, so it has a fast ligand or CD95 receptor, which is a death receptor and it also produces fast ligand, this both interact together and make the cell apoptosis. This is the two way how the peripheral tolerance occurs, leading to energy or apoptosis of T and B cells and one of the other mutation example is an autoimmune lymphoproliferative syndrome. Can you explain that, same like at all?
Peripheral Tolerance
Why Peripheral Tolerance Exists
- Central tolerance (thymus) is not perfect — some self-reactive T cells escape into the periphery
- Peripheral tolerance acts as the second safety net to silence or eliminate these cells
- Two main mechanisms: Anergy and Apoptosis (AICD)
Mechanism 1 — Anergy
- A T cell in the periphery encounters an antigen presented on MHC by a dendritic cell
- For full T cell activation, two signals are required:
- Signal 1: TCR binds antigen-MHC complex
- Signal 2: CD28 on the T cell binds B7 (CD80/CD86) on the dendritic cell
- B7 is only upregulated on dendritic cells during active inflammation / infection (danger signal)
- If a T cell recognizes a self-antigen in a non-inflammatory environment:
- Signal 1 fires (TCR binds)
- Signal 2 does NOT fire (no B7 upregulation, no danger signal)
- Result: T cell enters anergy — it is alive but permanently unresponsive, functionally silenced
- Anergic T cells cannot proliferate or produce cytokines even if stimulated again later
Mechanism 2 — Activation-Induced Cell Death (AICD) via Fas/FasL Pathway
- Also called the extrinsic apoptosis pathway
- When a T cell receives Signal 1 repeatedly without Signal 2, it upregulates two molecules on its own surface:
- Fas (CD95) — a death receptor
- FasL (CD95L) — the ligand for Fas
- The same cell (or a neighboring T cell) expresses both → Fas on one cell binds FasL on itself or adjacent cell
- This interaction activates caspase cascade → apoptosis
- This is called fratricidal killing or suicide depending on whether it is the same or neighboring cell
Correction on Your Note
- You called it "fast ligand" and "fast receptor" — the correct names are FasL (Fas Ligand) and Fas (CD95), also written as CD95 / CD95L
- Fas = the receptor (death receptor) → CD95
- FasL = the ligand → CD178 / CD95L
Additional Peripheral Tolerance Mechanisms (USMLE High-Yield)
- CTLA-4 — expressed on activated T cells; also binds B7 but with higher affinity than CD28, and delivers an inhibitory signal → brakes T cell activation
- Regulatory T cells (Tregs) — CD4⁺ CD25⁺ FoxP3⁺ cells that actively suppress autoreactive T cells via IL-10 and TGF-β
- PD-1 / PD-L1 pathway — PD-1 on T cells binds PD-L1 on peripheral tissues → suppresses T cell activity (exploited by tumors to evade immunity)
When Peripheral Tolerance Fails → Autoimmune Lymphoproliferative Syndrome (ALPS)
Mechanism
- Normally, autoreactive T cells that escape to periphery are eliminated by the Fas-FasL apoptosis pathway
- In ALPS, there is a mutation in Fas (CD95) or FasL
- Fas-FasL interaction fails → apoptosis does not occur → autoreactive lymphocytes accumulate and proliferate unchecked
Clinical Features of ALPS
- Massive non-malignant lymphadenopathy (lymph node enlargement)
- Splenomegaly (often massive)
- Hepatomegaly
- Autoimmune cytopenias — autoimmune hemolytic anemia, thrombocytopenia, neutropenia
- Elevated double-negative T cells (DNTs) — CD4⁻ CD8⁻ αβ T cells in peripheral blood — this is the diagnostic hallmark
- Increased risk of lymphoma (Hodgkin and non-Hodgkin)
Genetics
- Most commonly: FAS gene mutation (autosomal dominant, variable penetrance)
- Also: FasL, Caspase-10, Caspase-8 mutations
- Presents in childhood
Full Peripheral Tolerance Summary Flow
Now explain me about the antiphospholipid antibody. It is an antibody which is directed against proteins bound to the phospholipids and how this antiphospholipid antibody is important to include anticardiolipin false negatives and VDRL and RPR syphilis screening tests and what is anti-beta-2 glycoprotein 1 and lupus anticoagulant which will falsely elevate PTT.
antiphospholipid antibody syndrome pathophysiology thrombosis mechanism
anticardiolipin antibody false positive VDRL RPR syphilis antiphospholipid
lupus anticoagulant anti-beta-2 glycoprotein PTT prolonged antiphospholipid
Antiphospholipid Antibody Syndrome (APS)
What Are Antiphospholipid Antibodies?
- Antibodies directed not against phospholipids directly, but against proteins that are bound to phospholipids
- The phospholipid acts as a scaffold — the real antigenic target is the protein sitting on top of it
- Main protein target: β2-glycoprotein I (β2-GPI) bound to phospholipid membranes
- These antibodies exert a procoagulant effect → paradoxically cause clotting despite appearing as anticoagulants in lab tests
The Three Major Antiphospholipid Antibodies
1. Anti-β2-Glycoprotein I (Anti-β2-GPI)
- The most pathologically significant antibody
- β2-GPI is a plasma protein that naturally binds to phospholipid membranes (especially on platelets and endothelial cells)
- Normally β2-GPI inhibits coagulation — it is anticoagulant by nature
- Anti-β2-GPI antibody binds to the β2-GPI/phospholipid complex → neutralizes its anticoagulant function → tips the balance toward thrombosis
- Also activates platelets and endothelial cells directly → further procoagulant state
- Detected by ELISA
2. Lupus Anticoagulant (LA)
- Misleading name — it causes clotting in vivo, not anticoagulation
- It is called "anticoagulant" only because of what it does in the test tube (in vitro)
- Mechanism in vitro: LA antibody binds to phospholipids in the coagulation assay reagent → blocks phospholipid-dependent clotting steps (Factor X, prothrombin activation) → falsely prolongs PTT
- In vivo: binds phospholipids on platelets and endothelium → activates them → thrombosis
- Detected by: prolonged PTT that does NOT correct with mixing study (1:1 mix with normal plasma)
- If PTT corrects → factor deficiency (e.g., hemophilia)
- If PTT does not correct → inhibitor present → lupus anticoagulant
- Confirmed by: dilute Russell's viper venom time (dRVVT)
3. Anticardiolipin Antibody (aCL)
- Cardiolipin is a phospholipid found in mitochondrial membranes and also used as an antigen in syphilis screening tests
- Anti-cardiolipin antibodies (IgG, IgM, IgA) bind to the cardiolipin-β2-GPI complex
- Detected by ELISA
- IgG subtype most strongly associated with thrombosis and pregnancy loss
The Syphilis Connection — False Positive VDRL/RPR
- VDRL and RPR are non-treponemal screening tests for syphilis
- They work by detecting antibodies against a cardiolipin-lecithin-cholesterol antigen complex
- The reagent antigen is cardiolipin
- In APS, the patient already has anticardiolipin antibodies circulating in their blood
- These antibodies react with the cardiolipin in the VDRL/RPR reagent → false positive result
- The patient does NOT have syphilis
- How to confirm true syphilis vs false positive: use treponemal-specific tests — FTA-ABS or TPPA
- True syphilis → treponemal test positive
- APS false positive → treponemal test negative
Clinical Features of APS
- Arterial thrombosis — stroke, TIA, MI in young patients
- Venous thrombosis — DVT, pulmonary embolism (most common)
- Obstetric complications:
- Recurrent pregnancy loss (especially second/third trimester — due to placental thrombosis)
- Preeclampsia, IUGR
- Thrombocytopenia — mild, due to platelet consumption
- Livedo reticularis — mottled, net-like skin discoloration
- Libman-Sacks endocarditis — sterile, non-bacterial endocarditis (especially in SLE-associated APS)
Primary vs Secondary APS
- Primary APS — occurs alone, no underlying disease
- Secondary APS — occurs with an underlying autoimmune disease, most commonly SLE (up to 30–40% of SLE patients have antiphospholipid antibodies)
Diagnosis — Sapporo/Sydney Criteria
- Clinical: vascular thrombosis (arterial/venous) OR pregnancy morbidity
- Laboratory (any one of):
- Lupus anticoagulant positive
- Anticardiolipin IgG or IgM (medium/high titer)
- Anti-β2-GPI IgG or IgM (medium/high titer)
- Must be positive on two occasions at least 12 weeks apart (to exclude transient positivity)
Lab Summary Flow
High-Yield One-Liners
- APS antibodies target proteins bound to phospholipids, not phospholipids directly
- Lupus anticoagulant prolongs PTT in vitro, causes clots in vivo
- Anticardiolipin antibody causes false positive VDRL and RPR
- Confirm syphilis with FTA-ABS or TPPA (treponemal tests)
- Confirm lupus anticoagulant with mixing study (no correction) + dRVVT
- Recurrent second/third trimester pregnancy loss in a young woman → think APS
- APS + SLE = secondary APS; treat with heparin + aspirin in pregnancy, long-term warfarin for thrombosis
If PTT prolongation not corrected, then it is a lupus anticoagulant, which is present in the serum. If it is 1 to 1 mixed and PTT gets corrected, then it's factor deficiency. Can you explain it a little more detail?
PTT Mixing Study — Detailed Explanation
Why We Do a Mixing Study
- A prolonged PTT has two possible causes:
- Factor deficiency — not enough clotting factor to complete the intrinsic pathway
- Inhibitor present — something in the blood is actively blocking the coagulation pathway
- A mixing study distinguishes between these two by adding normal plasma (which contains all factors at 100%) to the patient's plasma
How the Mixing Study Works
- Take patient plasma (prolonged PTT) + normal plasma in a 1:1 ratio
- Normal plasma contains all clotting factors in full quantity
- Re-run the PTT on the mixed sample
Result Interpretation
If PTT Corrects After Mixing → Factor Deficiency
- Normal plasma donated its factors to the patient's plasma
- The missing factor is now present in sufficient quantity → coagulation proceeds normally → PTT normalizes
- The patient's plasma had no factors to give, but had nothing blocking the donated factors either
- Diagnoses: Hemophilia A (Factor VIII deficiency), Hemophilia B (Factor IX deficiency), Factor XI deficiency
If PTT Does NOT Correct After Mixing → Inhibitor Present
- Normal plasma donated its factors — but something in the patient's plasma destroyed or blocked those donated factors too
- The inhibitor is so potent it overpowers even the normal plasma's contribution
- This confirms an inhibitor is circulating in the patient's blood
Why Lupus Anticoagulant Prolongs PTT — The Mechanism
- The PTT test works by adding phospholipid reagent + activator + calcium to plasma and measuring how long clotting takes
- The phospholipid in the reagent acts as a surface on which Factor X and prothrombin activation complexes assemble
- Lupus anticoagulant binds and occupies these phospholipids in the reagent
- Without a free phospholipid surface, the coagulation complexes cannot assemble → reaction slows → PTT artificially prolonged
- This is purely a test tube artifact — the phospholipid in the reagent is not the same as what happens inside blood vessels
Why LA Causes Clotting In Vivo Despite Prolonging PTT In Vitro
- Inside the body, LA binds phospholipids on platelet surfaces and endothelial cell membranes
- β2-GPI sits on these phospholipid membranes acting as a natural anticoagulant
- LA neutralizes β2-GPI → removes the brake on coagulation → procoagulant state
- Also directly activates platelets and endothelial cells → further clot formation
- Result: arterial and venous thrombosis in vivo
Inhibitor Titre — Bethesda Assay
- If mixing study does not correct, the next step is to identify and quantify the inhibitor
- For Factor VIII inhibitor (e.g., acquired hemophilia): Bethesda assay — measures inhibitor units
- For lupus anticoagulant: confirmed by dilute Russell's viper venom time (dRVVT)
- dRVVT uses very low phospholipid concentration → LA has maximum effect → prolonged clotting time
- Then repeat with excess phospholipid → LA gets neutralized → time corrects
- Correction with excess phospholipid = confirms LA
Factor VIII Inhibitor vs Lupus Anticoagulant — Key Distinction
- Both prolong PTT and do not correct on mixing study
- Factor VIII inhibitor:
- Causes bleeding (destroys Factor VIII)
- Seen in hemophilia patients who developed antibodies after factor replacement, or in acquired hemophilia (middle-aged, autoimmune)
- Bethesda assay positive
- Lupus anticoagulant:
- Causes thrombosis (not bleeding)
- Seen in SLE, APS, infections, drugs
- dRVVT confirms
Full Mixing Study Algorithm Flow
got it
Like if you have to make an ultra-short note saying that anti-cardiolipin, false positive VDRL and RPR syphilis screening test, anti-beta-2-glycoprotein 1 a few points, and lupus anticoagulant falsely elevated PTT. But if it's mixed with 1% of normal serum, 1 ml of normal plasma with all factors including it to the patient's plasma. Though it's PTT elevated, then some inhibitor is present like anticardiolipin. If it's corrected and this PTT becomes normal, then it's a factor deficiency like hemophilias. So this as an ultra-short note I want all important highlighted points, ultra-short like for quick revision.
Antiphospholipid Antibodies — Quick Revision
Anticardiolipin Antibody
- Binds cardiolipin-β2GPI complex
- Cardiolipin is the antigen used in VDRL and RPR syphilis screening
- Anticardiolipin reacts with reagent → false positive VDRL/RPR
- Confirm true syphilis with FTA-ABS or TPPA (treponemal) → negative = false positive
Anti-β2-Glycoprotein I
- β2-GPI normally sits on phospholipid membranes and inhibits coagulation
- Anti-β2-GPI neutralizes it → removes anticoagulant brake → thrombosis
- Most pathologically significant antibody in APS
- Causes arterial/venous thrombosis + recurrent pregnancy loss
Lupus Anticoagulant
- Binds phospholipid in the PTT reagent → blocks coagulation complex assembly → PTT falsely prolonged
- In vitro = anticoagulant | In vivo = pro-thrombotic
- Confirmed by mixing study + dRVVT
PTT Mixing Study
- Mix patient plasma + normal plasma (1:1)
- Normal plasma has all factors at full levels
- Patient bleeding + no correction → Factor VIII inhibitor → Bethesda assay
- Patient clotting + no correction → Lupus anticoagulant → confirm with dRVVT