Atherosclerosis
Reading File
Finding Sources
Searching PubMed
"atherosclerosis"[MeSH Terms] AND ("pathogenesis" OR "treatment")
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
I now have very comprehensive content. Let me compile the complete answer.
Atherosclerosis
Definition
Atherosclerosis is an intimal-based disease of large and medium-sized arteries characterized by the formation of atheromatous plaques - raised lesions with a soft, grumous lipid core (mainly cholesterol and cholesterol esters with necrotic debris) covered by a fibrous cap. It is the single leading cause of morbidity and mortality in Western nations, responsible for roughly half of all deaths, and underlies coronary artery disease, stroke, aortic aneurysm, and peripheral vascular disease.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 467
Epidemiology
While risk reduction and improved therapies have sharply decreased atherosclerosis-related mortality in high-resource nations over recent decades, adoption of Western diet and lifestyle in lower-resource countries has driven sharp increases there. Death rates for coronary artery disease in Africa, India, and Southeast Asia now exceed those in the United States; Eastern European countries have rates 3-5 times higher than the United States and 7-12 times higher than Japan.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 467
Risk Factors
Risk factors identified through prospective studies (e.g., the Framingham Heart Study) include:
| Non-Modifiable (Constitutional) | Modifiable |
|---|---|
| Genetic variation / family history | Hyperlipidemia (elevated LDL, low HDL, elevated Lp(a)) |
| Increasing age | Hypertension |
| Male sex | Cigarette smoking |
| Diabetes mellitus | |
| Obesity / metabolic syndrome | |
| Physical inactivity | |
| High-sensitivity CRP (inflammation markers) | |
| Homocysteinemia |
Multiple risk factors have greater-than-additive effects. Familial predisposition is usually polygenic; Mendelian disorders (e.g., familial hypercholesterolemia) account for only a small fraction.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 468
Pathogenesis: The Response-to-Injury Hypothesis
The central concept is that atherosclerosis is a vascular response to endothelial injury and chronic inflammation. Key sequential steps:
- Endothelial injury / dysfunction - increased permeability, enhanced leukocyte adhesion, altered gene expression
- Lipoprotein accumulation - LDL and oxidized LDL enter the vessel wall
- Monocyte adhesion and migration - monocytes adhere to dysfunctional endothelium via adhesion molecules, cross into the intima, and differentiate into macrophages
- Foam cell formation - macrophages ingest oxidized LDL via scavenger receptors → lipid-laden foam cells → fatty streak (earliest visible lesion)
- Platelet adhesion and growth factor release
- SMC recruitment and proliferation - smooth muscle cells migrate from the media into the intima, proliferate, and produce ECM
- Extracellular lipid accumulation and necrosis → necrotic core
- Calcification of ECM and necrotic debris in late stages → "hardening of the arteries"
Three Key Causes of Endothelial Dysfunction
-
Hemodynamic disturbances: Plaques preferentially occur at branch points, vessel ostia, and the posterior abdominal aorta where turbulent, non-laminar flow predominates. Laminar flow induces the transcription factor KLF2 (Krüppel-like factor-2) which upregulates atheroprotective genes; turbulent flow reverses this (notably, statins also work in part via KLF2 upregulation).
-
Hypercholesterolemia: Elevated LDL, reduced HDL, and elevated Lp(a). Oxidized LDL is particularly atherogenic - it stimulates monocyte adhesion, is toxic to endothelium, and is taken up by macrophage scavenger receptors (bypassing normal feedback regulation).
-
Inflammation: Systemic inflammation (elevated CRP, fibrinogen) and local cytokine production sustain the atherogenic process.
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 469-471
Diagrams of Pathogenesis
Monocyte adhesion → foam cell formation:
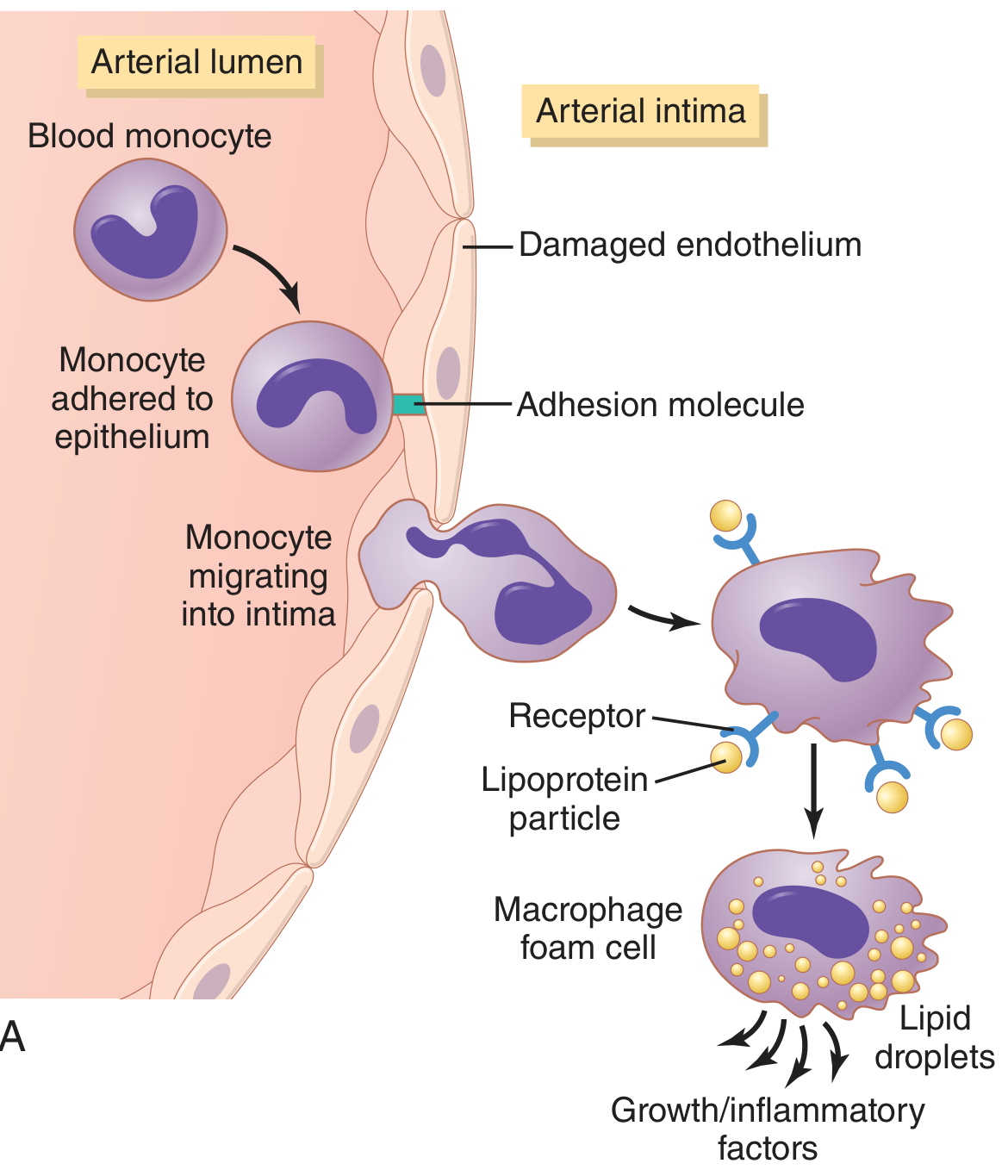
Fig. 69.8A from Guyton & Hall: Monocyte adhesion via adhesion molecules, migration into the intima, and conversion to macrophage foam cells releasing growth/inflammatory factors.
Full progression - healthy artery to fibrofatty atheroma:
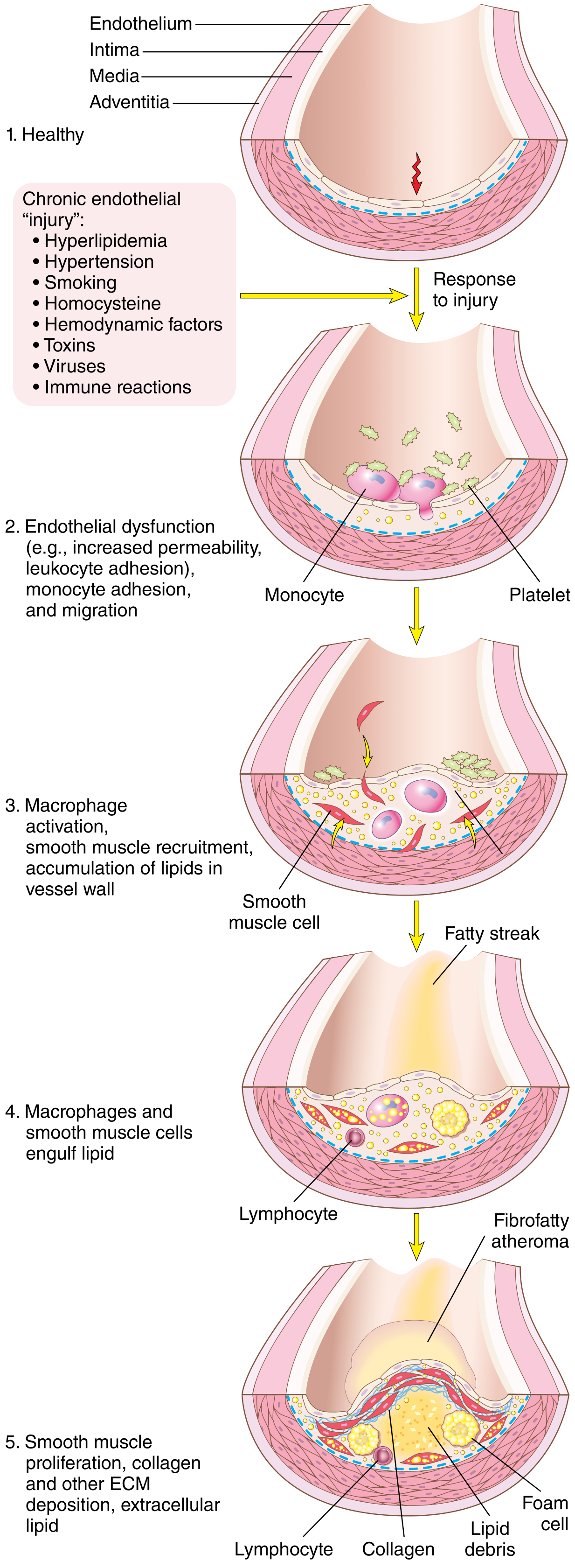
Fig. 11.8 from Robbins Cotran: Five stages from healthy vessel to fibrofatty atheroma with SMC proliferation, ECM deposition, and extracellular lipid accumulation.
Plaque growth to rupture and thrombosis:
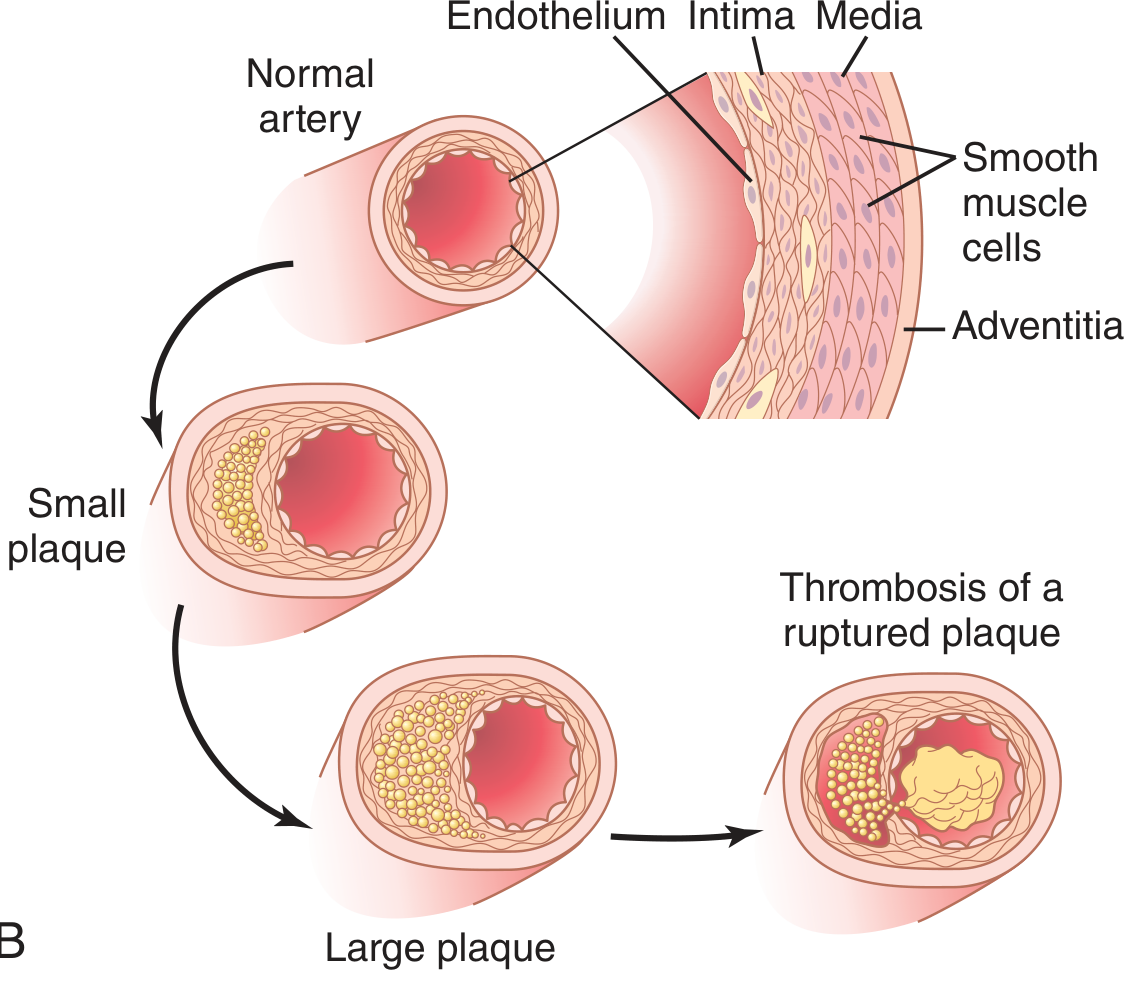
Fig. 69.8B from Guyton & Hall: Progression from small plaque → large plaque → plaque rupture with thrombosis.
Morphology of the Atherosclerotic Plaque
A fully developed atheromatous plaque consists of:
- Fibrous cap: Dense collagen, scattered SMCs, macrophages, and T lymphocytes
- Necrotic core: Cholesterol crystals, lipid debris, dead foam cells, calcifications
- Shoulder regions: More cellular - SMCs, macrophages, T cells (structurally weakest zones)
- Neovascularization: Small vessels grow into the plaque, potentially contributing to intraplaque hemorrhage
Plaque Distribution
Large elastic arteries (aorta, carotid, iliac) and large-to-medium muscular arteries (coronary, popliteal) are the primary sites. In any given artery the abdominal aorta is more affected than the thoracic aorta.
Stable vs. Vulnerable (Unstable) Plaques
| Feature | Stable Plaque | Vulnerable Plaque |
|---|---|---|
| Fibrous cap | Thick, dense | Thin |
| Lipid core | Small | Large |
| Inflammation | Minimal | Dense (macrophages, T cells) |
| Calcification | Often present | Variable |
| Risk | Chronic ischemia by stenosis | Rupture → acute thrombosis |
The "Glagov phenomenon": early plaques cause outward remodeling of the media to preserve lumen size, so luminal narrowing is a late event. Critical stenosis occurs when the plaque produces a 70-75% reduction in luminal cross-sectional area, enough to cause ischemia at rest or with minimal exertion.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 473
Complications of Atherosclerotic Plaques
- Plaque rupture / fissuring - the most dangerous complication; exposes the thrombogenic necrotic core to blood, triggering acute thrombosis (cause of most MIs and strokes)
- Luminal thrombosis - may be occlusive (infarct) or non-occlusive (mural thrombus)
- Atheroembolism - plaque debris or thrombus breaks off and occludes distal vessels (e.g., "cholesterol emboli" to kidneys, retina, toes)
- Calcification - leads to arterial stiffness ("hardening of the arteries")
- Aneurysm formation - medial ischemia (from impaired diffusion through thick intima + inflammation) weakens the wall, particularly in the abdominal aorta
Clinical Manifestations
| Territory | Manifestation |
|---|---|
| Coronary arteries | Angina, unstable angina, myocardial infarction, sudden cardiac death |
| Cerebral/carotid arteries | TIA, ischemic stroke |
| Aorta | Abdominal aortic aneurysm |
| Renal arteries | Renovascular hypertension, ischemic nephropathy |
| Mesenteric arteries | Intestinal ischemia |
| Lower limb arteries | Claudication, rest pain, gangrene |
Prevention and Treatment
Lifestyle modifications: Smoking cessation, Mediterranean diet, aerobic exercise, weight loss, blood pressure control, glycemic control.
Pharmacotherapy:
- Statins (HMG-CoA reductase inhibitors): First-line for LDL reduction; also have pleiotropic anti-inflammatory effects (including via KLF2 upregulation). Recent meta-analysis (JAMA Cardiol, 2025) showed that alternative LDL-lowering strategies combining lower-intensity statins + ezetimibe achieved comparable cardiovascular outcomes to high-intensity statin monotherapy.
- Ezetimibe: Reduces intestinal cholesterol absorption; used add-on to statins.
- PCSK9 inhibitors (evolocumab, alirocumab): Markedly reduce LDL; used in statin-intolerant patients or very high-risk cases.
- Antihypertensives: ACE inhibitors, ARBs have vascular-protective effects beyond BP lowering.
- Antiplatelet agents: Aspirin, clopidogrel for secondary prevention.
- GLP-1 receptor agonists: Emerging evidence for cardiovascular risk reduction.
Summary Points
- Atherosclerosis = endothelial injury + inflammation + lipid accumulation in the intima
- The fatty streak is the earliest lesion; mature plaques take decades to develop
- Stable plaques cause gradual stenosis; vulnerable plaques rupture and cause acute events
- Roughly half of deaths in Western nations are attributable to atherosclerotic disease
- Risk factor modification (especially LDL lowering) significantly reduces events
Sources: Robbins & Kumar Basic Pathology, p. 314 | Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 467-474 | Guyton and Hall Textbook of Medical Physiology, p. 851
This is a shared conversation. Sign in to Orris to start your own chat.