Give me structured comprehensive answer for only pathogenesis of tb for md exams 15marks
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content and diagrams needed. Let me compose a comprehensive, exam-structured answer on TB pathogenesis.
Pathogenesis of Tuberculosis
Source: Robbins, Cotran & Kumar — Pathologic Basis of Disease; Robbins & Kumar Basic Pathology
1. Introduction
Tuberculosis (TB) is a chronic granulomatous disease caused by Mycobacterium tuberculosis, a slender, acid-fast bacillus with a lipid-rich cell wall. The pathogenesis is fundamentally a story of host–pathogen interaction: the same cell-mediated immune response that provides protection also produces the characteristic tissue damage (caseating granulomas, cavitation). The outcome of infection depends entirely on the interplay between bacterial virulence and host immunity.
2. Causative Organism
| Feature | Detail |
|---|---|
| Organism | M. tuberculosis hominis (most cases); M. bovis (bovine milk, rare) |
| Staining | Acid-fast (Ziehl-Neelsen / carbol fuchsin) |
| Cell wall | Mycolic acids, lipoarabinomannan (LAM), cord factor (trehalose 6,6′-dimycolate) |
| Virulence | Intracellular survival by blocking phagolysosome fusion |
3. Transmission
- Route: Inhalation of airborne droplet nuclei (1–5 μm) from an individual with active pulmonary TB
- Infectious dose: As few as 1–3 bacilli can initiate infection
- Target: Alveolar macrophages in the distal airspaces (lower lobe upper segment / upper lobe lower segment)
4. Pathogenesis — Step-by-Step Sequence
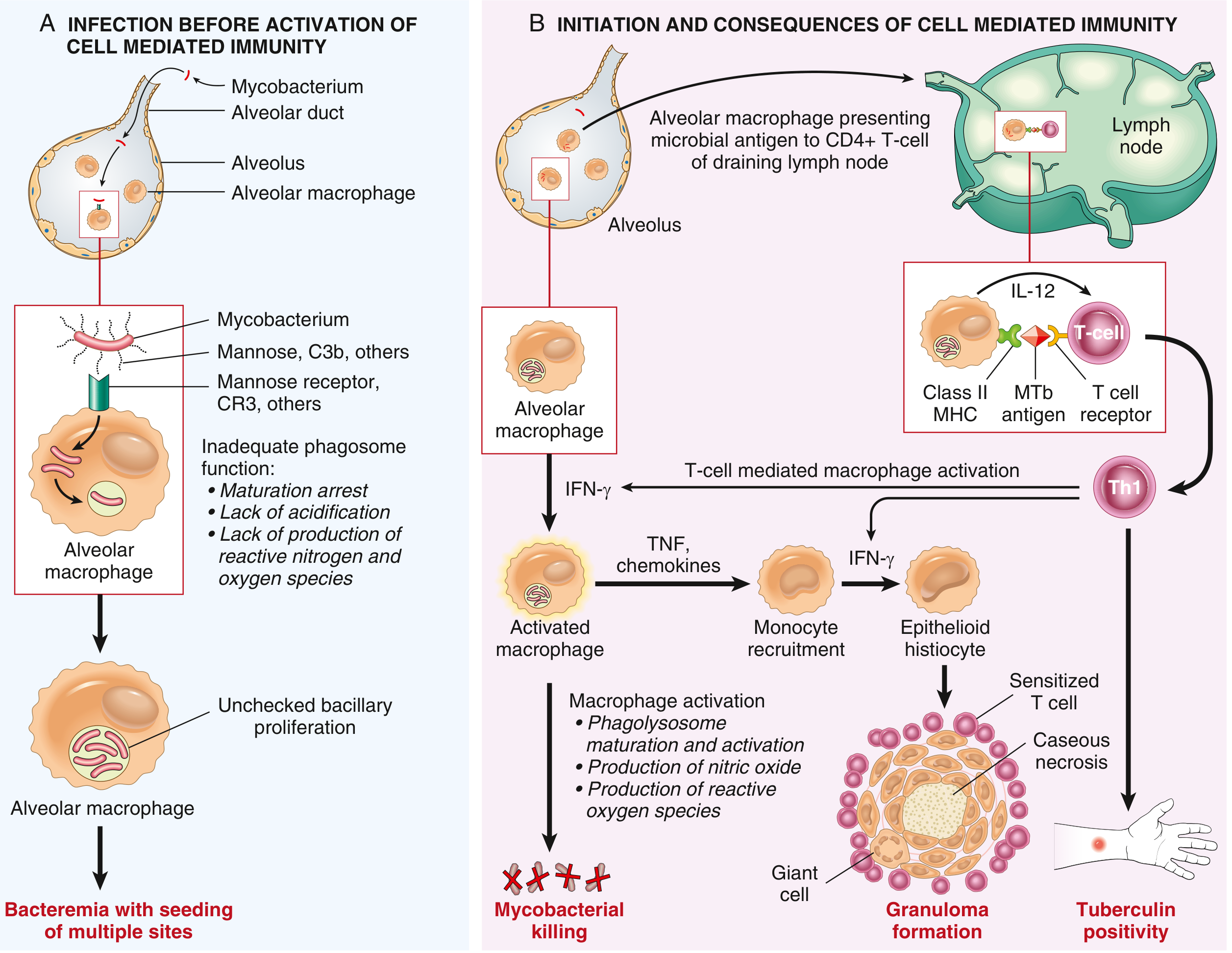
Phase A: Before Activation of Cell-Mediated Immunity (First 3 weeks)
Step 1 — Entry into Macrophages
- Inhaled bacilli are engulfed by alveolar macrophages via receptor-mediated phagocytosis
- Key receptors: mannose receptor (binds mycobacterial cell wall mannose residues), complement receptor 3 (CR3/CD11b) (binds C3b-opsonized bacteria), and scavenger receptors
- Dendritic cells are also infected and carry organisms to draining lymph nodes
Step 2 — Intracellular Survival and Replication
- M. tuberculosis arrests phagosomal maturation and blocks phagolysosome fusion
- Mechanism: The bacterium recruits host protein coronin to the phagosome membrane → coronin activates the phosphatase calcineurin → inhibits phagosome-lysosome fusion
- Result: Bacteria replicate unchecked within phagocytic vacuoles, protected from lysosomal microbicidal mechanisms
- During these first 3 weeks, bacilli proliferate in alveolar macrophages and air spaces → bacteremia with seeding of multiple organs
- Clinically: most patients are asymptomatic or have a mild flu-like illness
Step 3 — Innate Immune Recognition (PAMPs & TLRs)
- Mycobacterial lipoarabinomannan (LAM) → binds TLR2
- Unmethylated CpG nucleotides → bind TLR9
- These interactions trigger production of pro-inflammatory cytokines, initiating the bridge to adaptive immunity
Phase B: Adaptive (Cell-Mediated) Immunity (From ~3 weeks)
Step 4 — Antigen Presentation and Th1 Differentiation
- Infected dendritic cells migrate from the lung to draining hilar lymph nodes
- Mycobacterial antigens are presented to CD4+ T cells via MHC class II
- Macrophages secrete IL-12 and IL-18 → drive differentiation of naïve CD4+ T cells into Th1 cells
- TLR2 stimulation by mycobacterial ligands promotes IL-12 production
Step 5 — Th1-Mediated Macrophage Activation and Bacterial Killing
Th1 cells produce IFN-γ — the master cytokine of TB immunity:
| IFN-γ Effect | Mechanism |
|---|---|
| Phagolysosome activation | Overcomes coronin block → acidification of phagosome |
| Nitric oxide (NO) | iNOS activation → reactive nitrogen intermediates kill bacteria |
| Reactive oxygen species (ROS) | Oxidative burst in activated macrophages |
| Autophagy | Sequesters and destroys intracellular bacteria |
| Antimicrobial peptides | Mobilizes defensins (cathelicidin) |
Activated macrophages also secrete TNF and chemokines → recruit additional monocytes to the site.
Importance of TNF: Patients on anti-TNF therapy (rheumatoid arthritis, IBD) have markedly increased risk of TB reactivation.
Step 6 — Granuloma Formation (The Hallmark Lesion)
- IFN-γ activates macrophages → transform into epithelioid histiocytes (large, pale, eosinophilic cells with abundant cytoplasm)
- Epithelioid cells aggregate into a granuloma; some fuse to form Langhans giant cells (peripheral horseshoe/arc arrangement of nuclei) or foreign-body giant cells
- Caseous necrosis develops at the center — a creamy white, cheese-like material representing liquefied cell debris and a hypersensitivity reaction
- Granulomas are surrounded by a collar of lymphocytes and fibroblasts
Step 7 — Tuberculin (Delayed Hypersensitivity)
- The same Th1 response produces delayed-type hypersensitivity (Type IV)
- Detectable by the tuberculin skin test (Mantoux/PPD) or IGRA (in-vitro IFN-γ release assay)
- A positive test signals T-cell sensitization — NOT necessarily active disease
- Tuberculin conversion appears at ~3–8 weeks post-infection
- Loss of tuberculin positivity in an infected patient is an ominous sign of waning immunity
5. Primary Tuberculosis (Ghon Complex)
After the Th1 response is mounted, the primary infection is contained in ~95% of immunocompetent individuals:
| Component | Description |
|---|---|
| Ghon focus | 1–1.5 cm area of gray-white caseating consolidation in the distal airspace (lower upper lobe or upper lower lobe, subpleural) |
| Lymphangitis | Tracking of bacilli along lymphatic channels |
| Hilar lymphadenopathy | Caseous hilar/mediastinal lymph nodes |
| Ghon complex | Ghon focus + lymphangitis + regional lymph node caseation |
| Ranke complex | Healed Ghon complex with fibrosis and calcification (seen on X-ray) |
Fate of the Ghon complex (95% of cases):
- Progressive fibrosis → calcification (Ranke complex)
- Organisms become latent (dormant in macrophages for decades)
- No further disease unless immunity wanes
Progressive primary TB (5% of cases):
- Occurs in overtly immunocompromised patients (HIV+ with CD4 <200, severe malnutrition, infants)
- Resembles acute lobar pneumonia — lower lobe consolidation, hilar lymphadenopathy, pleural effusion
- May progress to miliary TB (hematogenous dissemination) or tuberculous meningitis
6. Latent TB
- After primary infection, viable but non-replicating organisms persist in macrophages (especially in the lung apex — higher oxygen tension favors dormancy)
- Patient is asymptomatic, non-infectious
- Lifetime risk of reactivation: ~5–10% in immunocompetent individuals
- Risk rises to 10–16% per year in HIV-infected patients
7. Secondary (Post-primary / Reactivation) Tuberculosis
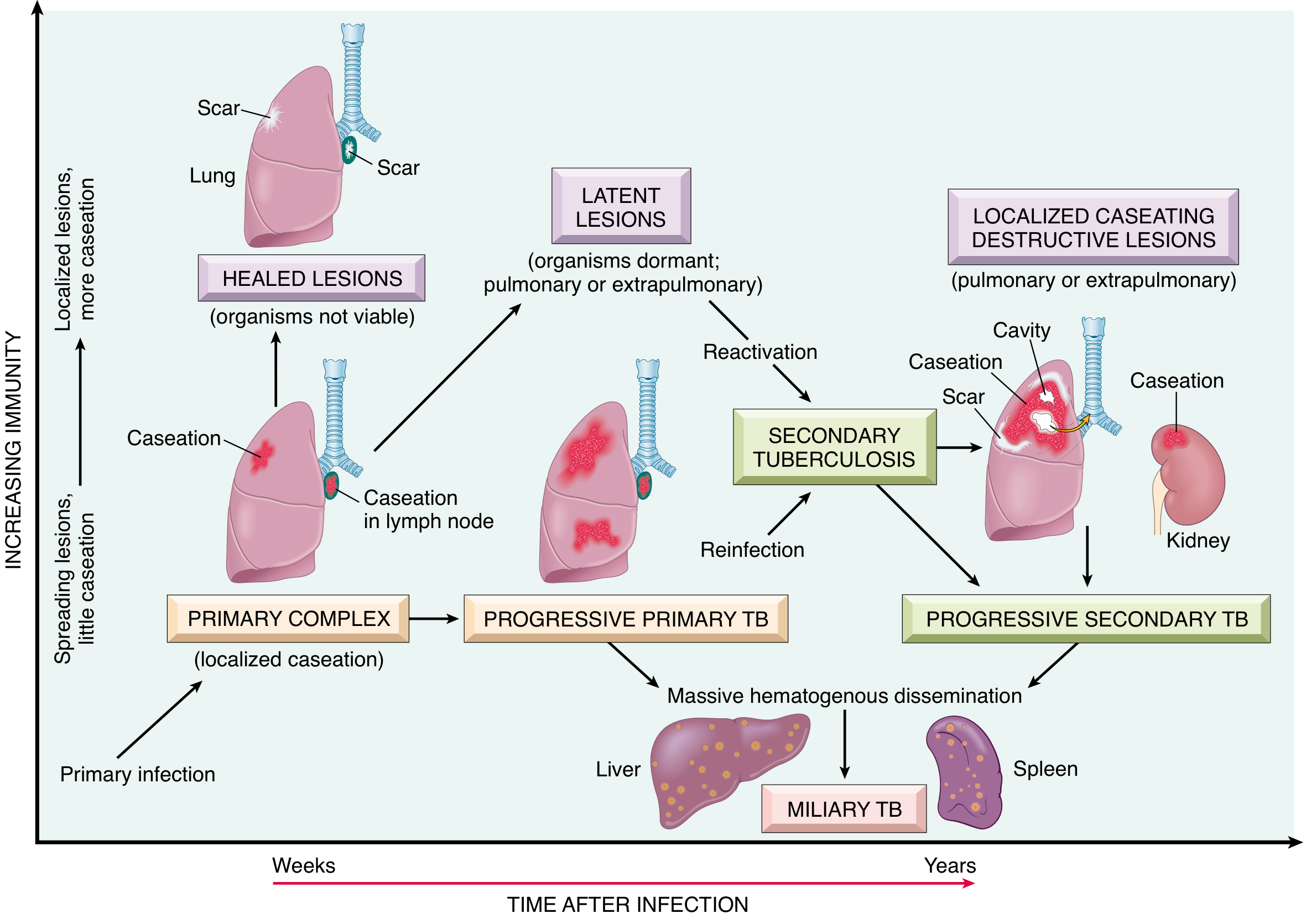
- Occurs in a previously sensitized host: either reactivation of latent infection or exogenous reinfection
- Due to preexisting hypersensitivity, the bacilli elicit a prompt, intense tissue response — rapid walling off of the focus
- Location: Classically the apex of the upper lobe (high O₂ tension favors bacterial growth)
- Regional lymph nodes are less prominently involved (unlike primary TB) due to rapid hypersensitivity response
Key Differences: Primary vs. Secondary TB
| Feature | Primary TB | Secondary TB |
|---|---|---|
| Host | Previously unexposed | Previously sensitized |
| Source | Exogenous (new infection) | Endogenous (reactivation) or exogenous (reinfection) |
| Location | Mid-lung / lower lobe | Lung apex (upper lobe) |
| Lymphadenopathy | Prominent hilar | Minimal |
| Cavitation | Rare | Common |
| Bacteremia | Common (initial) | Less common |
| Ghon complex | Present | Absent (prior fibrocalcific scar) |
8. Host Susceptibility Factors
| Factor | Mechanism |
|---|---|
| HIV/AIDS | Depletion of CD4+ Th1 cells → granulomas absent → poorly contained infection |
| Anti-TNF therapy | Impairs macrophage activation and granuloma integrity → reactivation |
| Corticosteroids / immunosuppressants | Suppress Th1 response |
| Diabetes mellitus | Impaired neutrophil and macrophage function |
| Silicosis | Silica particles disrupt macrophage function |
| Malnutrition | Impairs cell-mediated immunity |
| Renal failure | Uremia-related immune deficiency |
| Genetic mutations | Loss of IL-12Rβ1, IFN-γR, TLR → severe susceptibility |
9. Summary of Key Cytokines and Molecules
| Molecule | Source | Role |
|---|---|---|
| IL-12 | Macrophages/DCs | Drives Th1 differentiation |
| IFN-γ | Th1 CD4+ T cells | Activates macrophages; central effector cytokine |
| TNF-α | Activated macrophages | Monocyte recruitment, granuloma maintenance |
| Nitric oxide (NO) | Activated macrophages | Mycobacterial killing |
| IL-18 | APCs | Co-stimulates Th1 with IL-12 |
| Coronin | Host cell | Recruited by MTB to block phagolysosome fusion |
10. Summary Flow Chart
Inhalation of M. tuberculosis
↓
Alveolar macrophage phagocytosis
(Mannose receptor + CR3)
↓
Phagolysosome fusion blocked (coronin/calcineurin)
Unchecked intracellular replication
↓ (first 3 weeks)
Bacteremia → seeding of organs (asymptomatic/flu-like)
↓
Dendritic cells migrate to hilar lymph nodes
Antigen presentation (MHC II) to CD4+ T cells
↓
IL-12/IL-18 → Th1 differentiation
↓
IFN-γ → macrophage activation
• iNOS → NO → mycobacterial killing
• Autophagy → intracellular clearance
• Phagolysosome → maturation restored
↓
TNF + chemokines → epithelioid histiocytes
→ Granuloma + caseous necrosis
↓
OUTCOME:
95% → Ghon complex → fibrosis/calcification (Ranke) → Latent TB
5% → Progressive primary TB → Miliary TB / Meningitis
↓ (years later, if immunity wanes)
Secondary TB → Apical cavitation → Active disease
Key Points for 15-Mark Exam
- Virulence mechanism = phagolysosome fusion block via coronin-calcineurin pathway
- Central cytokine = IFN-γ (from Th1 cells) — activates macrophages to kill bacilli
- Tissue damage = same immune response (Th1 hypersensitivity) that protects also causes caseous necrosis
- Ghon complex = Ghon focus + lymphangitis + hilar lymphadenopathy (triad)
- Ranke complex = healed, calcified Ghon complex
- 95% rule = 95% of primary infections are contained and become latent
- Reactivation = upper lobe apex, rapid hypersensitivity response, cavitation
- TNF antagonists → reactivation of latent TB (clinically important)
- HIV → loss of CD4+ Th1 cells → absent granulomas → disseminated, progressive disease
- Tuberculin positivity = evidence of sensitization (not disease); tuberculin negativity in infected patient = ominous sign of failing immunity
Sources: Robbins, Cotran & Kumar — Pathologic Basis of Disease (10th ed.), pp. 350–353; Robbins & Kumar Basic Pathology (11th ed.), pp. 475–478
This is a shared conversation. Sign in to Orris to start your own chat.