Describe and discuss microbiology and Immunology of viral hepatitis -
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive data from authoritative sources. Let me compile the complete answer.
Microbiology and Immunology of Viral Hepatitis
Viral hepatitis refers to inflammation of the liver caused by a group of hepatotropic viruses - HAV, HBV, HCV, HDV, and HEV - each with distinct virological properties, transmission routes, and immunological interactions. Other viruses (EBV, CMV, yellow fever) can also cause hepatitis, but the five classical agents are the primary focus here.
Overview Comparison Table
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Family | Picornaviridae | Hepadnaviridae | Flaviviridae | Deltaviridae | Hepeviridae |
| Genus | Hepatovirus | Orthohepadnavirus | Hepacivirus | Deltavirus | Hepevirus |
| Genome | ssRNA (7.5 kb) | Partial dsDNA (3.2 kb) | ssRNA (9.4 kb) | Circular defective ssRNA (1.7 kb) | ssRNA (7.2 kb) |
| Virion size | 27-32 nm, icosahedral | 42 nm (Dane particle) | 60 nm, spherical | 35 nm, spherical | 30-32 nm, icosahedral |
| Envelope | No | Yes (HBsAg) | Yes | Yes (uses HBsAg) | No |
| Transmission | Fecal-oral | Parenteral/perinatal/sexual | Parenteral | Parenteral | Fecal-oral |
| Chronic disease | Never | 5-10% adults; 95% neonates | >80% | 10% co-inf; >80% superinfection | Never (except immunocompromised) |
| Oncogenic | No | Yes | Yes | Unknown | No |
| Fulminant | Rare (0.1%) | Rare | Rare | Frequent | High in pregnancy (~20%) |
(Jawetz Medical Microbiology 28e; Robbins & Kumar Basic Pathology)
1. Hepatitis A Virus (HAV)
Microbiology
HAV is a 27-32 nm, non-enveloped, icosahedral, positive-sense single-stranded RNA virus belonging to the genus Hepatovirus within the family Picornaviridae. Its genome is 7.5 kb. Only one serotype is known, with no antigenic cross-reactivity with other hepatitis viruses. Genomic sequence analysis divides HAV into seven genotypes based on the 1D/2A gene junction.
HAV is remarkably stable - resistant to 20% ether, acid (pH 1.0 for 2 hours), and heat (60°C for 1 hour). It can survive for months dried at 25°C. Inactivation requires autoclaving (121°C for 20 min), boiling for 5 minutes, UV irradiation, or formalin/chlorine treatment. Foods must be heated above 85°C for at least 1 minute.
Transmission is fecal-oral via contaminated water, food (especially shellfish), and close personal contact. The virus is shed in stool for 2-3 weeks before and 1 week after the onset of jaundice - the window of maximum infectivity. Incubation is 2-6 weeks.
Immunology and Pathogenesis
HAV itself does not appear to be directly cytopathic to hepatocytes. Liver injury is mediated primarily by the cellular immune response, particularly cytotoxic CD8+ T lymphocytes targeting infected hepatocytes.
Serological course:
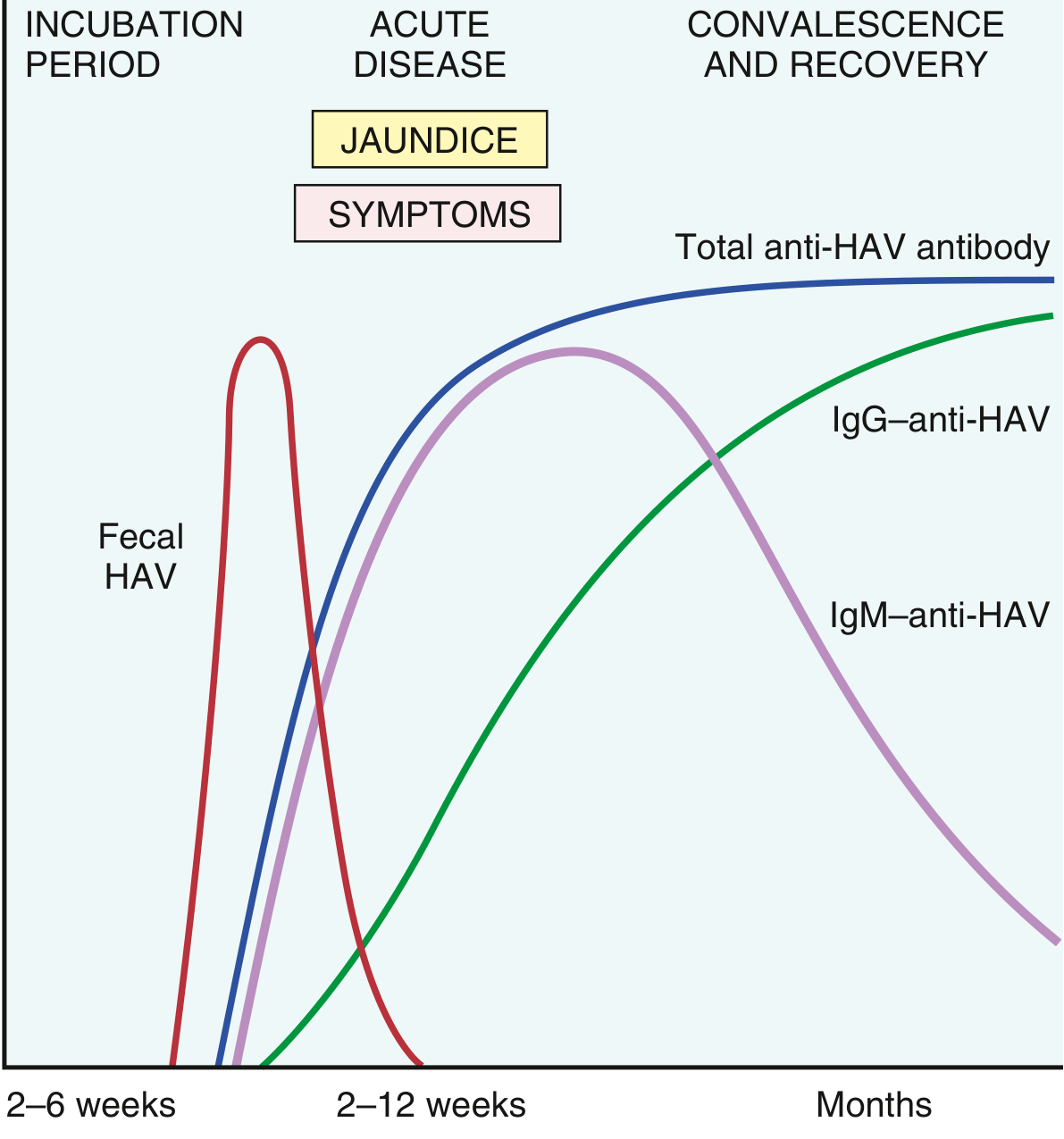
- IgM anti-HAV appears at the onset of symptoms and is the reliable marker of acute infection (positive up to 4-6 months post-infection). Fecal virus shedding ends as IgM rises.
- IgG anti-HAV follows, persisting for years and conferring lifelong immunity.
- HAV viremia is transient, making bloodborne transmission very rare.
Outcomes: Disease is more severe in adults (75-90% icteric) than in children (mostly subclinical). Chronic hepatitis never develops. Fulminant hepatitis is rare (~0.1%). Complete recovery occurs in >98%. An effective vaccine (available since 1995) has reduced US cases by over 95%.
2. Hepatitis B Virus (HBV)
Microbiology and Structure
HBV is the prototype hepadnavirus (family Hepadnaviridae), classified as the only human DNA hepatitis virus. It has a partial dsDNA genome of 3.2 kb in a circular configuration with a single-stranded gap region.
Electron microscopy of HBsAg-positive serum reveals three morphological forms:
- 22 nm spherical particles - the most numerous; composed exclusively of HBsAg; represent excess surface antigen
- Tubular/filamentous forms - same 22 nm diameter but up to 200 nm long; also HBsAg only
- 42 nm spherical Dane particles - the complete, infectious virion; the outer envelope carries HBsAg surrounding a 27 nm nucleocapsid core containing HBcAg and the viral DNA polymerase
Key antigens and their clinical significance:
| Marker | Significance |
|---|---|
| HBsAg | Surface antigen; first marker to appear; indicates active infection |
| HBeAg | Secreted form of HBcAg; marker of high infectivity and active viral replication |
| HBcAg | Core antigen; not detectable in serum (intracellular only) |
| Anti-HBs | Protective antibody; indicates recovery or successful vaccination |
| Anti-HBc IgM | Acute infection marker; also detectable in "window period" |
| Anti-HBc IgG | Past or chronic infection; not protective alone |
| Anti-HBe | Appears as HBeAg clears; indicates low replication |
| HBV DNA | Gold standard for viral load and replication activity |
HBV replication involves a reverse transcriptase step - the pregenomic RNA intermediate is reverse-transcribed into the DNA genome. This is the target of nucleoside/nucleotide analogues (tenofovir, entecavir).
HBV is not directly cytopathic; liver damage is immune-mediated.
Immunology and Pathogenesis of HBV
The outcome of HBV infection is determined by the host immune response:
Successful immune response (adults, >90%):
- CD8+ cytotoxic T cells (CTLs) recognize HBcAg/HBeAg epitopes on infected hepatocytes and kill them
- CD4+ T helper cells coordinate the response
- Neutralizing anti-HBs antibodies clear circulating virions
- This leads to acute, self-limited hepatitis with complete viral clearance
Failed immune response (neonates, immunosuppressed) → chronic HBV:
- Neonatal infection leads to tolerance to HBV antigens - the immature immune system fails to mount a CTL response
- 95% of neonates infected perinatally become chronic carriers vs. only 5-10% of adult-infected individuals
- HBV persists as covalently closed circular DNA (cccDNA) in the nucleus, which serves as a template for transcription and is resistant to current antivirals
Chronic HBV histology: Hepatocytes show characteristic "ground glass" appearance on H&E staining (accumulation of HBsAg in ER), confirmed by specific immunostaining:
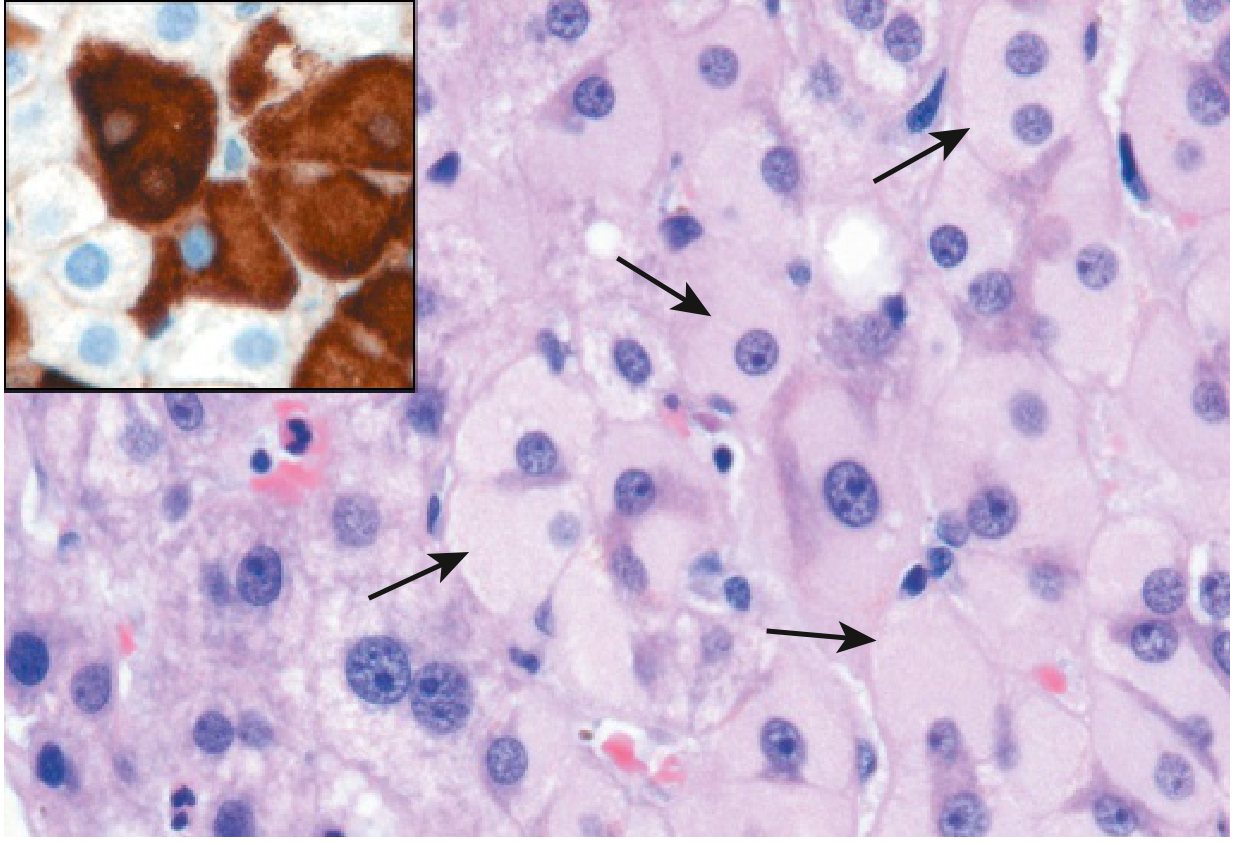
Natural history phases of chronic HBV:
- Immune tolerant phase - high viral replication, normal ALT, minimal hepatic inflammation
- Immune active/clearance phase - active CTL attack, elevated ALT, hepatic necroinflammation
- Inactive carrier phase - HBeAg seroconversion to anti-HBe, low HBV DNA, near-normal ALT
- HBeAg-negative chronic hepatitis - reactivation with pre-core/basal core promoter mutant virus
Extrahepatic immune manifestations of HBV include polyarteritis nodosa (immune complex deposition), membranous glomerulonephritis, and cryoglobulinemia - all mediated by circulating HBsAg-anti-HBs immune complexes.
HBV and hepatocellular carcinoma (HCC): HBV integrates into the host genome and can activate oncogenes (e.g., cyclin A) and disrupt tumor suppressor genes. Chronic necroinflammation drives regeneration → cirrhosis → HCC. The HBx protein from HBV is also a transcriptional transactivator implicated in carcinogenesis.
3. Hepatitis C Virus (HCV)
Microbiology
HCV is an enveloped, positive-sense ssRNA virus (9.4 kb), a member of the Flaviviridae family, genus Hepacivirus. Its genome encodes a single polyprotein that is cleaved by both host and viral proteases into 10 functional proteins, including:
- NS3/4A protease - processes the polyprotein (target of protease inhibitors: simeprevir, grazoprevir)
- NS5A protein - essential for viral assembly (target of NS5A inhibitors: ledipasvir, daclatasvir)
- NS5B RNA-dependent RNA polymerase - replicates the genome (target of polymerase inhibitors: sofosbuvir)
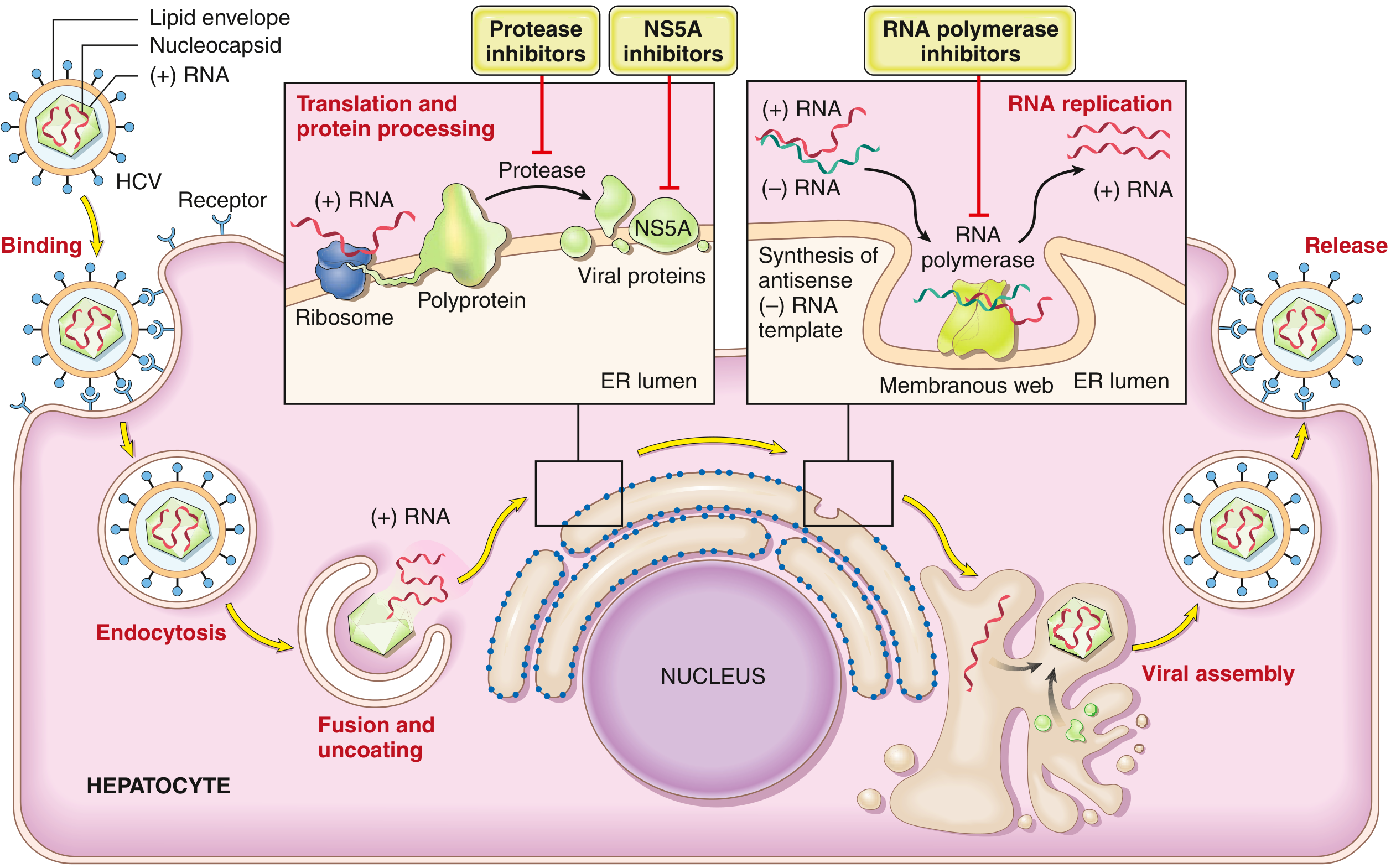
Because the NS5B polymerase has very low fidelity, new genetic variants arise rapidly. There are seven major HCV genotypes worldwide. Within a single infected patient, the virus exists as a population of closely related variants called quasispecies - a key mechanism by which HCV evades immune elimination.
Immunology and Pathogenesis of HCV
Mechanisms of immune evasion (why HCV becomes chronic in >80%):
- Quasispecies variation - constant antigenic drift outpaces B and T cell responses
- NS3/4A protease cleaves TRIF and MAVS - two critical intracellular signaling adaptors for innate antiviral interferon responses, blocking IFN-β production
- Impaired NK cell and dendritic cell function in chronic infection
- T cell exhaustion - chronic antigen stimulation leads to upregulation of inhibitory receptors (PD-1, Tim-3, CTLA-4) on HCV-specific CD8+ T cells, impairing their function
- Regulatory T cells (Tregs) suppress anti-HCV immune responses
Serological course:
- HCV RNA is detectable in blood 1-3 weeks after exposure
- Serum transaminases (ALT) rise concurrently
- Anti-HCV antibodies (detected by ELISA) appear later (weeks to months) and are not protective
- In ~85% of cases, acute infection is asymptomatic
- Chronic infection (defined as HCV RNA persisting >6 months) occurs in 80-90% of individuals
- In chronic infection, HCV RNA persists in 90% of patients despite circulating neutralizing antibodies
Pathological hallmark of chronic HCV: Portal tract expansion with a dense lymphoid infiltrate (often with lymphoid follicle formation) is characteristic:
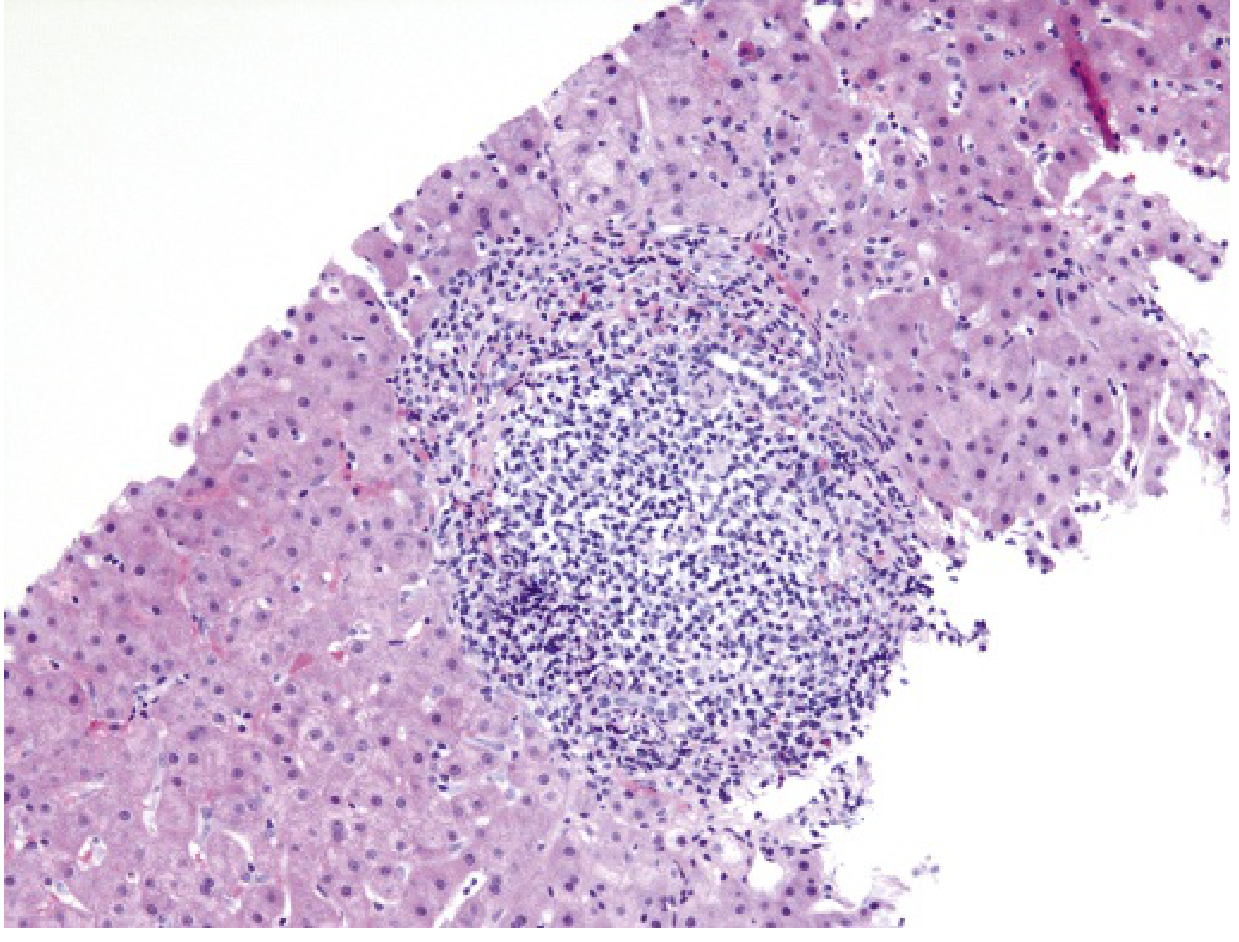
Progression: ~20% of chronically infected individuals develop cirrhosis over 20-30 years. Progression is accelerated by older age, male gender, alcohol, HBV/HIV coinfection, obesity, and metabolic syndrome. Cirrhosis is a major risk factor for HCC (HCV accounts for ~1/3 of liver cancer cases in the US).
Treatment: The development of direct-acting antivirals (DAAs) targeting the viral protease, NS5A, and polymerase has produced SVR (sustained virological response, i.e., cure) rates >95% with 8-12 week courses, revolutionizing HCV management.
4. Hepatitis D Virus (HDV)
Microbiology
HDV (delta agent) is a unique, defective RNA virus - it can only infect individuals who are also infected with HBV, because it relies on HBsAg to form its envelope. The HDV virion is 35-37 nm, enveloped (using HBsAg), with an inner ribonucleoprotein core consisting of the 1.7 kb circular, negative-sense ssRNA genome and the hepatitis D antigen (HDAg).
HDAg exists in two forms:
- Small HDAg (p24) - promotes viral RNA replication
- Large HDAg (p27) - inhibits replication and is required for virion assembly
Immunology and Clinical Patterns
HDV infection occurs in two clinical settings:
- Co-infection (HDV + HBV simultaneously): Usually self-limited, resembles acute hepatitis B. Both viruses typically cleared together. Higher risk of fulminant hepatitis (especially in IV drug users).
- Superinfection (HDV infecting a chronic HBV carrier): More severe - causes acute exacerbation or severe chronic hepatitis. Chronic HDV infection follows in >80% of superinfections. Accelerates progression to cirrhosis and HCC.
Serology:
- HDAg detectable in serum during early acute infection
- IgM anti-HDV - reliable marker of recent HDV exposure; short-lived in co-infection, persistent in superinfection
- In superinfection: high, persistent IgG and IgM anti-HDV, persistent HBsAg and HDV RNA
- Coinfection is confirmed by the simultaneous presence of IgM anti-HDAg and IgM anti-HBcAg
Prevention: Since HDV cannot exist without HBV, HBV vaccination fully prevents HDV infection.
Estimated 15 million individuals worldwide are infected with HDV. Highest prevalence in the Amazon basin, Africa, the Middle East, and Southern Italy.
5. Hepatitis E Virus (HEV)
Microbiology
HEV is a non-enveloped, icosahedral, positive-sense ssRNA virus (7.2 kb), the sole member of the genus Hepevirus, family Hepeviridae. The virion is 30-32 nm. Four major genotypes exist - genotypes 1 and 2 infect only humans; genotypes 3 and 4 are zoonotic (reservoirs: pigs, monkeys, cats, deer, rodents).
Transmission: Fecal-oral, primarily via contaminated water. Epidemics occur in Asia, Indian subcontinent (HEV accounts for 30-60% of sporadic acute hepatitis in India), sub-Saharan Africa, and Mexico. Sporadic zoonotic cases occur in Europe and North America (linked to pig farming and consumption of undercooked pork/game).
Incubation is 4-5 weeks.
Immunology
- IgM anti-HEV and IgG anti-HEV, plus PCR for HEV RNA, are the diagnostic tools
- Infection is usually self-limiting - no chronic disease in immunocompetent hosts
- In immunocompromised patients (solid-organ transplant recipients, HIV-infected), chronic HEV infection can occur
- Pregnant women (especially in 3rd trimester) are at greatly increased risk of fulminant hepatic failure: mortality approaches 20% - the mechanism involves hormonal modulation of immune responses (elevated estrogen/progesterone impairs T cell and innate immune control)
- No specific antiviral treatment; ribavirin is used in chronic/severe cases
Summary: Key Immunological Mechanisms
| Aspect | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Direct cytopathicity | No | No | Limited | Limited | No |
| Key effector | CD8+ CTL | CD8+ CTL, anti-HBs | CD8+ CTL | CD8+ CTL (via HBV) | CD8+ CTL |
| Acute marker | IgM anti-HAV | HBsAg, IgM anti-HBc | HCV RNA | IgM anti-HDV + IgM anti-HBc | IgM anti-HEV |
| Protective antibody | IgG anti-HAV | Anti-HBs | None effective | Anti-HBs (prevents HBV) | IgG anti-HEV |
| Immune evasion | Transient viremia | Neonatal tolerance, cccDNA | Quasispecies, T cell exhaustion, MAVS/TRIF cleavage | Depends on HBV | - |
| Chronic mechanism | N/A | Immune tolerance, cccDNA | T cell exhaustion, quasispecies | Requires HBV, immune dysregulation | N/A |
Pathological Features of Chronic Viral Hepatitis
All chronic hepatitis viruses produce broadly similar histological patterns that vary by grade (activity) and stage (fibrosis):
- Lobular hepatitis - hepatocyte necrosis/apoptosis (acidophil/Councilman bodies)
- Interface hepatitis (piecemeal necrosis) - periportal hepatocyte destruction by lymphocytes
- Portal tract infiltration - predominantly lymphocytes, plasma cells
- Bridging necrosis - portoportal or portocentral necrosis (marks severe activity)
- Fibrosis → cirrhosis - stellate cell activation by inflammatory cytokines (TGF-β)
HCV-specific: dense portal lymphoid aggregates/follicles, steatosis (especially genotype 3), mild bile duct damage
HBV-specific: ground-glass hepatocytes (HBsAg accumulation in ER)
Sources: Jawetz, Melnick & Adelberg's Medical Microbiology 28e (pp. 517-527); Robbins & Kumar Basic Pathology (pp. 599-606); Tietz Textbook of Laboratory Medicine 7e
Describe and discuss microbiology and Immunology of viral hepatitis in exam point of view with detailed explanation
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive, exam-level content from authoritative sources. Let me compile the complete, exam-focused answer with all images and detailed explanations.
🎓 Microbiology and Immunology of Viral Hepatitis — Exam-Focused Complete Guide
❗ HIGH-YIELD EXAM MNEMONIC: "ABC DE — Hepatitis 5"
| Letter | Virus | Key Fact |
|---|---|---|
| A | HAV | Acute only, Alimentary (fecal-oral) |
| B | HBV | Blood-borne, Big threat (chronicity, cancer) |
| C | HCV | Chronic king (>80%), Cancer risk |
| D | HDV | Dependent on HBV (Defective virus) |
| E | HEV | Enteric (fecal-oral), Expectant mothers danger |
SECTION 1: MASTER COMPARISON TABLE (Must Memorize for Exams)
| Feature | HAV | HBV | HCV | HDV | HEV |
|---|---|---|---|---|---|
| Common Name | "Infectious hepatitis" | "Serum hepatitis" | "Non-A Non-B" | "Delta agent" | "Enteric Non-A Non-B" |
| Family | Picornaviridae | Hepadnaviridae | Flaviviridae | Deltaviridae | Hepeviridae |
| Genus | Hepatovirus | Orthohepadnavirus | Hepacivirus | Deltavirus | Hepevirus |
| Genome | (+) ssRNA, 7.5 kb | Partial dsDNA, 3.2 kb | (+) ssRNA, 9.4 kb | Circular (-) ssRNA, 1.7 kb | (+) ssRNA, 7.2 kb |
| Virion Size | 27-32 nm, icosahedral | 42 nm spherical (Dane particle) | 60 nm spherical | 35-37 nm spherical | 30-32 nm, icosahedral |
| Envelope | No (naked) | Yes (HBsAg) | Yes | Yes (borrows HBsAg) | No (naked) |
| Transmission | Fecal-oral | Parenteral, sexual, perinatal | Parenteral (blood > sexual) | Parenteral | Fecal-oral (waterborne) |
| Onset | Abrupt | Insidious | Insidious | Abrupt | Abrupt |
| Incubation | 15-50 days (2-6 wks) | 45-160 days (mean 8 wks) | 14-180 days (mean 9 wks) | 15-64 days | 15-50 days (4-5 wks) |
| Chronicity | NEVER | 5-10% adults; 90-95% neonates | >80% | 10% coinfection; >80% superinfection | Never (except immunocompromised) |
| Fulminant disease | Rare (<0.1%) | Rare (0.1-0.5%) | Rare | Common | 20% in pregnancy |
| Carrier state | No | Yes | Yes | Yes | No |
| Oncogenic | No | Yes (HCC, cirrhosis) | Yes (HCC, cirrhosis) | Unknown | No |
| Vaccine available | Yes | Yes | No | Prevented by HBV vaccine | No (in development) |
| Mortality | <0.5% | 1-2% | ~4% | High-very high | Normal: 1-2%; Pregnant: 20% |
(Jawetz Medical Microbiology 28e; Robbins & Kumar Basic Pathology; Medical Microbiology 9e)
SECTION 2: HEPATITIS A VIRUS (HAV)
2.1 Microbiology / Virology
Exam Trigger: "Smallest hepatitis virus," "Picornavirus," "No envelope," "One serotype"
- Classification: Picornaviridae family, genus Hepatovirus
- Structure: 27-32 nm, non-enveloped, icosahedral capsid, positive-sense ssRNA (7.5 kb)
- Single serotype exists (clinically important - one vaccine covers all strains)
- Seven genotypes based on the 1D/2A gene junction; no cross-reactivity with other hepatitis viruses
- Stability - Exam important:
- Stable to: 20% ether, acid (pH 1.0 for 2 hrs), heat 60°C for 1 hour
- Destroyed by: Autoclaving (121°C/20 min), boiling 5 min, UV light, formalin 1:4000, chlorine 10-15 ppm
- Food must be heated >85°C for 1 minute to inactivate
- Cell culture: grows in primate cell lines; no cytopathic effect usually seen
- Detected originally in stool by immune electron microscopy
2.2 Transmission and Epidemiology
- Route: Fecal-oral (contaminated water, food, shellfish, infected food handlers)
- Maximum infectivity: Virus shed in stool 2-3 weeks BEFORE and 1 week AFTER jaundice onset
- Viremia is transient - bloodborne transmission extremely rare; blood not specifically screened
- Endemic in countries with poor sanitation infrastructure
2.3 Immunology and Pathogenesis
Key Exam Concept: HAV is NOT directly cytopathic - damage is IMMUNE-MEDIATED
Mechanism of liver injury:
- HAV infects hepatocytes after fecal-oral entry
- CD8+ cytotoxic T lymphocytes (CTLs) are the principal effectors of hepatocyte destruction
- CTLs recognize viral antigens presented on MHC class I of infected hepatocytes and lyse them
- This immune-mediated lysis causes the transaminase elevation and jaundice
Serological course - MUST KNOW:
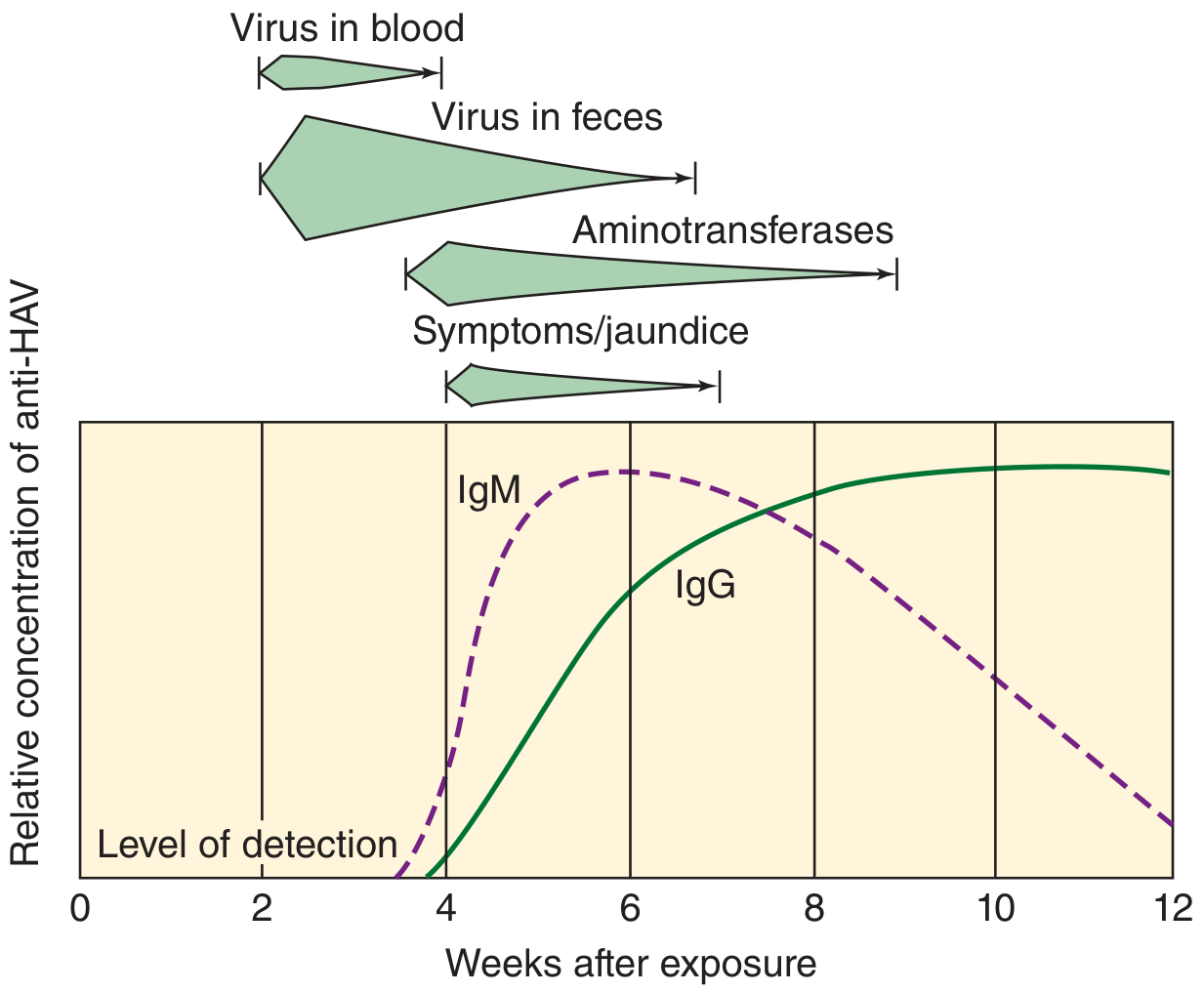
| Marker | Timing | Meaning |
|---|---|---|
| Fecal HAV | 2 wks before symptoms | Peak infectivity window |
| IgM anti-HAV | Onset of symptoms; lasts 3-6 months | Diagnostic marker of ACUTE infection |
| IgG anti-HAV | Shortly after disease onset; lasts decades | Marker of past infection / immunity |
| Aminotransferases (ALT/AST) | Rise with symptoms | Marker of hepatocyte damage |
ELISA is the method of choice for measuring HAV antibodies.
2.4 Disease Outcomes
| Children | Adults | |
|---|---|---|
| Subclinical infection | 80-95% | 10-25% |
| Icteric disease | 5-20% | 75-90% |
| Complete recovery | >98% | >98% |
| Chronic disease | None | None |
| Mortality | 0.1% | 0.3-2.1% |
- Disease is more severe in adults than children
- Relapses can occur 1-4 months after initial resolution
- Vaccine (available since 1995) has reduced US cases by >95%
SECTION 3: HEPATITIS B VIRUS (HBV)
3.1 Microbiology / Virology
Exam Trigger: "Only DNA hepatitis virus," "Dane particle," "Reverse transcriptase," "cccDNA"
Classification: Hepadnaviridae (from HEPatitis + ADNA + virus), genus Orthohepadnavirus
Unique Feature: ONLY human hepatitis virus with a DNA genome
3.2 Structure - Three Morphological Forms in Serum (EXAM FAVORITE)
On electron microscopy of HBsAg-positive serum, three distinct forms are seen:
| Form | Size | Composition | Quantity | Infectious? |
|---|---|---|---|---|
| 1. Small spheres | 22 nm | HBsAg only | Most numerous | NO |
| 2. Tubular/filamentous | 22 nm diameter, up to 200 nm long | HBsAg only | Abundant | NO |
| 3. Dane particle | 42 nm | Complete virion: HBsAg envelope + 27 nm HBcAg nucleocapsid + dsDNA + polymerase | Rare | YES |
Exam Note: Small spheres and tubules represent excess HBsAg overproduction - important diagnostic and immunological targets. Antibody against HBsAg (anti-HBs) is the only NEUTRALIZING/PROTECTIVE antibody.
3.3 Genome Organization (3.2 kb, Circular Partial dsDNA)
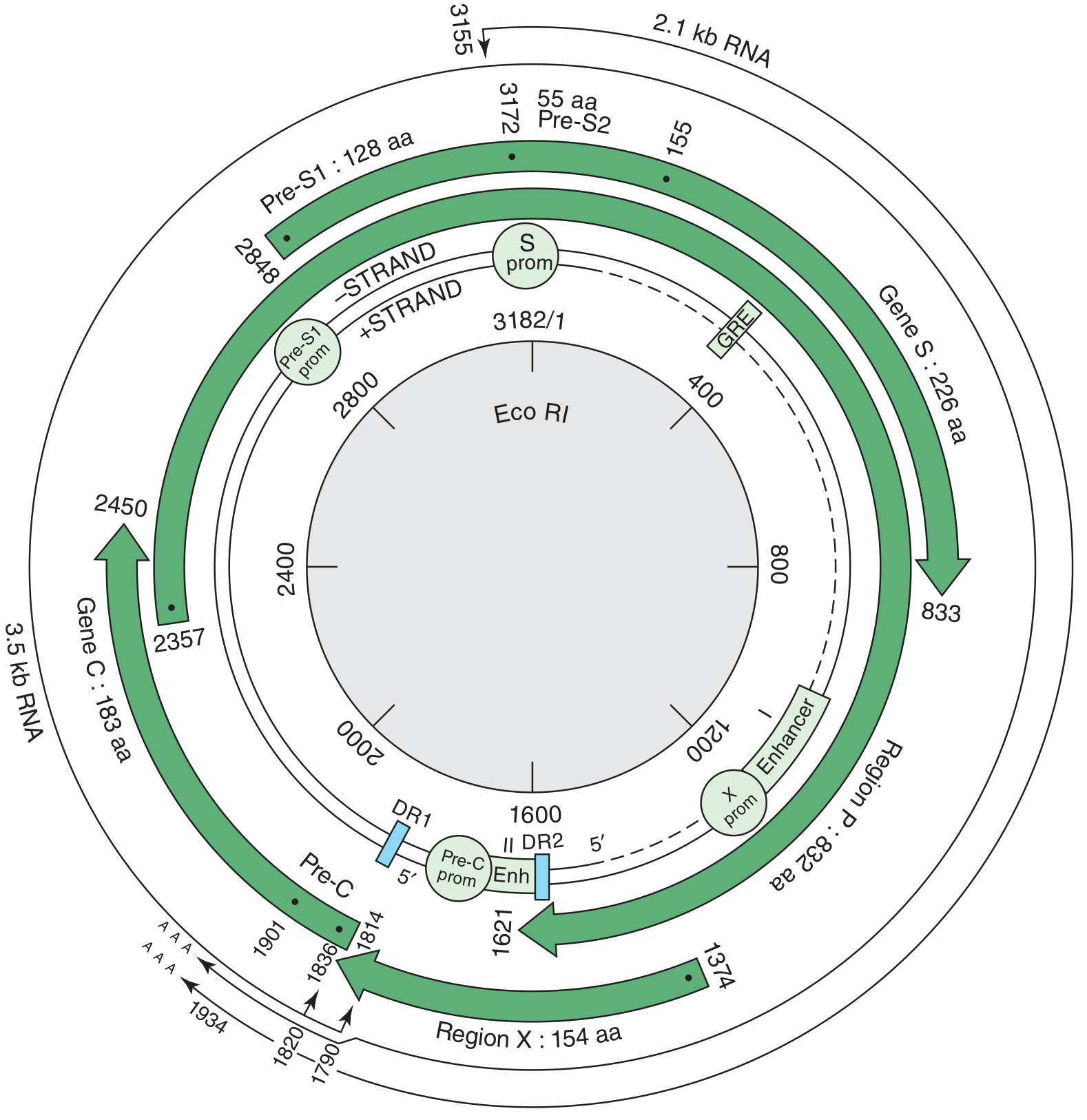
Four Open Reading Frames (ORFs) - Must Know:
| Gene | Protein Product | Function |
|---|---|---|
| S gene (Pre-S1, Pre-S2, S) | HBsAg (surface antigen) | Forms viral envelope; basis of vaccine; 3 subtypes of HBsAg: adw, ayw, adr, ayr (common subtype in US = adw) |
| C gene (Pre-C, C) | HBcAg (core antigen) + HBeAg (pre-core product) | HBcAg forms nucleocapsid; HBeAg is secreted = marker of HIGH REPLICATION |
| P gene | DNA Polymerase (with reverse transcriptase activity) | Replication of genome via RNA intermediate; target of antivirals (tenofovir, entecavir) |
| X gene | HBx protein | Transcriptional transactivator; required for viral replication; implicated in hepatocellular carcinoma |
3.4 Replication Cycle (EXAM HIGH YIELD)
The unique feature of HBV replication: DNA → RNA → DNA (uses reverse transcriptase - like a retrovirus!)
Steps:
- Virion attaches to hepatocyte; NTCP (sodium taurocholate cotransporting polypeptide) is the receptor
- Uncoating; partial dsDNA genome enters nucleus
- In nucleus: converted to cccDNA (covalently closed circular DNA) - the "persistence reservoir" - not eliminated by antivirals
- cccDNA transcribed → pregenomic RNA (pgRNA, 3.5 kb) + mRNAs
- pgRNA encapsidated with HBcAg and viral polymerase
- Within capsid: reverse transcription of pgRNA → minus-strand DNA; then plus-strand DNA synthesis begins (incomplete)
- Immature cores bud from pre-Golgi membranes, acquiring HBsAg envelope
- Released as virions OR recycled back to nucleus to maintain cccDNA pool
Key Point: cccDNA in hepatocyte nucleus is the reason HBV cannot be cured by current antivirals - it serves as a transcriptional template even during treatment.
3.5 Viral Antigens and Antibodies - COMPLETE SEROLOGY (MOST TESTED TOPIC)
Interpretation Table:
| Marker | HBsAg | Anti-HBs | HBeAg | Anti-HBe | IgM anti-HBc | Total anti-HBc | HBV DNA |
|---|---|---|---|---|---|---|---|
| Acute infection | + | - | + | - | + | + | + |
| Window period | - | - | - | - | + | + | - |
| Resolved infection | - | + | - | + | - | + | - |
| Chronic infection (active) | + | - | + | - | - | + | + |
| Chronic (inactive carrier) | + | - | - | + | - | + | low/- |
| Vaccination | - | + | - | - | - | - | - |
Serology in Acute and Chronic HBV - Key Diagram:
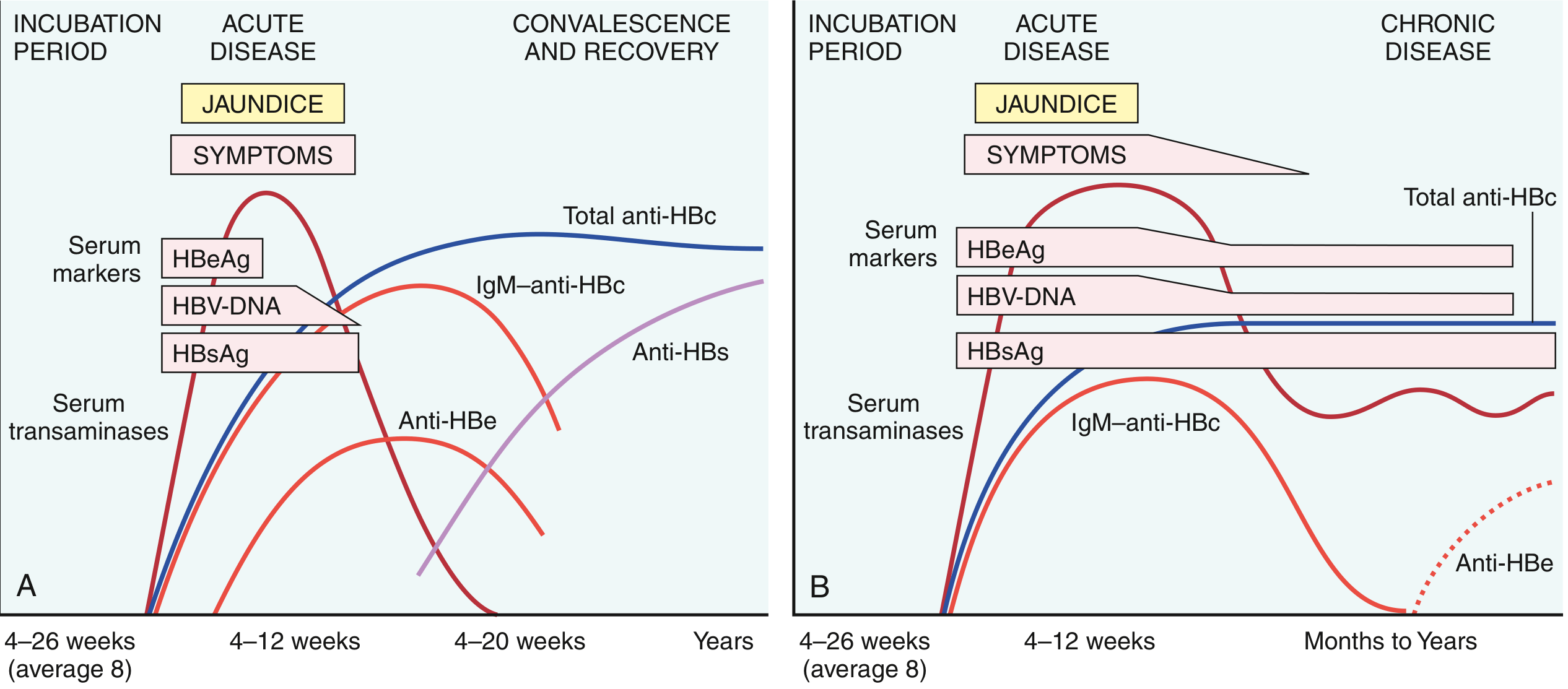
Individual marker meanings - EXAM POINTS:
| Marker | Exam Significance |
|---|---|
| HBsAg | First marker to appear; indicates ACTIVE infection (acute or chronic); basis of blood screening |
| Anti-HBs | ONLY protective/neutralizing antibody; confers immunity; seen after recovery OR vaccination |
| HBeAg | Marker of HIGH replication and HIGH INFECTIVITY; secreted form of core protein |
| Anti-HBe | Signals waning of acute infection; low replication |
| IgM anti-HBc | Marker of ACUTE infection; ALSO present during window period (only detectable marker!) |
| IgG anti-HBc | Marker of past or chronic infection; NOT protective alone |
| HBcAg | NOT detectable in serum (intracellular only - liver biopsy needed) |
| HBV DNA | Gold standard for viral replication; used to monitor treatment response |
| "Window period" | HBsAg has cleared but Anti-HBs not yet present; ONLY IgM anti-HBc detectable |
3.6 HBV Immunology and Pathogenesis
Key Exam Concept: HBV is NOT directly cytopathic - liver damage is entirely immune-mediated
Innate immune response (early):
- IFN-α and IFN-β produced by infected hepatocytes and dendritic cells
- NK cells recruited
- These limit but cannot eradicate HBV
Adaptive immune response (determining outcome):
- CD4+ T helper cells recognize HBcAg/HBeAg peptides on MHC class II of APCs → produce IFN-γ, IL-2
- CD8+ cytotoxic T cells (CTLs) recognize HBcAg, HBsAg on MHC class I of infected hepatocytes → direct killing + IFN-γ secretion
- B cells → anti-HBs (neutralizing), anti-HBe, anti-HBc
Why does chronic infection develop?
| Factor | Mechanism |
|---|---|
| Neonatal infection | Immature immune system cannot mount effective CTL response; HBeAg crosses placenta and induces central tolerance to HBcAg/HBeAg → virus "invisible" to T cells |
| cccDNA persistence | Not eliminated by immune response or antivirals; maintains viral transcription |
| T cell exhaustion | Chronic antigen exposure upregulates PD-1, CTLA-4, Tim-3 on HBV-specific CTLs → functional exhaustion |
| Regulatory T cells | Tregs suppress anti-HBV CTL activity |
| Viral immune evasion | Pre-core mutants stop producing HBeAg → escape anti-HBe immunity |
Age-dependent chronicity rates (EXTREMELY HIGH YIELD):
| Age at infection | Chronicity rate |
|---|---|
| Neonates/infants (perinatal) | 90-95% |
| Young children (1-5 yrs) | 25-30% |
| Older children | 10% |
| Adults | 5-10% |
Mnemonic: "The younger, the worse" - immature immunity = chronic disease; mature immunity = resolution
Natural History Phases of Chronic HBV:
| Phase | HBsAg | HBeAg | HBV DNA | ALT | Liver Damage |
|---|---|---|---|---|---|
| 1. Immune tolerant | + | + | Very high | Normal | Minimal |
| 2. Immune active (clearance) | + | + | High | Elevated | Active hepatitis |
| 3. Inactive carrier | + | - (anti-HBe +) | Low | Normal | Minimal |
| 4. HBeAg-negative reactivation | + | - | Moderate-high | Elevated | Active hepatitis |
Extrahepatic immune-complex manifestations of HBV:
- Polyarteritis nodosa - HBsAg-anti-HBs immune complexes deposit in vessel walls
- Membranous glomerulonephritis - immune complex deposition in glomeruli
- Cryoglobulinemia, serum sickness-like prodrome in acute infection
HBV and Hepatocellular Carcinoma (HCC):
- HBx protein transactivates host oncogenes
- HBV DNA integration into host genome → chromosomal instability
- Cyclin A activation, tumor suppressor gene disruption
- Chronic inflammation → regenerative hyperplasia → cirrhosis → HCC
- Risk is independent of cirrhosis (HBV can cause HCC directly)
SECTION 4: HEPATITIS C VIRUS (HCV)
4.1 Microbiology / Virology
Exam Trigger: "Most common cause of chronic viral hepatitis worldwide," "Quasispecies," "No protective vaccine," "NS5B polymerase"
- Classification: Flaviviridae family, genus Hepacivirus
- Structure: Enveloped, positive-sense ssRNA, 9.4 kb genome
- Single large polyprotein (~3000 aa) cleaved into 10 proteins by host and viral proteases
HCV Proteins - Key for Exams:
| Protein | Type | Function | Drug Target? |
|---|---|---|---|
| Structural | |||
| Core | Nucleocapsid | Virus assembly | No |
| E1, E2 | Envelope glycoproteins | Receptor binding (CD81, SR-BI, CLDN1, OCLN) | Partial |
| Non-structural | |||
| NS3/4A | Serine protease | Polyprotein processing; cleaves MAVS and TRIF (immune evasion) | YES - Protease inhibitors (glecaprevir, grazoprevir) |
| NS5A | Phosphoprotein | Viral assembly; RNA replication complex | YES - NS5A inhibitors (ledipasvir, velpatasvir, pibrentasvir) |
| NS5B | RNA-dependent RNA polymerase | Genome replication | YES - Polymerase inhibitors (sofosbuvir) |
Seven major genotypes (1 through 7); genotype 1 most common in US/Europe; genotype 3 associated with steatosis
Quasispecies concept (HIGH YIELD):
- NS5B polymerase has very low fidelity (no proofreading)
- Thousands of genetic variants accumulate in a single patient
- Each patient harbors a quasispecies - a cloud of related variants
- This is the key reason why HCV evades immune control and why no vaccine is possible
4.2 HCV Life Cycle
4.3 HCV Immunology - Why Chronicity Dominates
Exam Concept: HCV has MULTIPLE sophisticated immune evasion strategies
Mechanisms of immune evasion leading to chronic infection (>80%):
| Mechanism | Detail |
|---|---|
| 1. Quasispecies variation | Continuous antigenic drift; B and T cells can never keep up; neutralizing antibodies ineffective |
| 2. NS3/4A protease cleaves MAVS | Mitochondrial Antiviral Signaling protein - adaptor for RIG-I; cleavage blocks IFN-β production |
| 3. NS3/4A protease cleaves TRIF | Adaptor for TLR3; blocking this further impairs innate IFN response |
| 4. CD8+ T cell exhaustion | Chronic antigen drives upregulation of PD-1, Tim-3, LAG-3, CTLA-4; CTLs become functionally impaired |
| 5. NK cell dysfunction | Altered NK cell cytotoxicity and cytokine production |
| 6. Regulatory T cells (Tregs) | Suppress HCV-specific immune responses |
| 7. Impaired dendritic cell function | Reduced antigen presentation and IFN production |
Serological course of HCV:
| Marker | Acute resolved | Chronic |
|---|---|---|
| HCV RNA | Detectable 1-3 wks after exposure; then clears | Persists (detectable in 90% despite antibodies) |
| ALT | Elevated (episodic) | Fluctuating (episodic elevations) |
| Anti-HCV | Appears weeks-months later; persists | Persists |
| Anti-HCV protects? | NO - not neutralizing | NO |
Diagnosis:
- ELISA for anti-HCV: screening (but can't distinguish acute from chronic or resolved)
- PCR for HCV RNA: confirms active infection; used to monitor treatment
- Genotyping: important for treatment decisions
4.4 Clinical Outcomes
- 85% of acute infections are asymptomatic
- 80-90% progress to chronic hepatitis (highest chronicity of all hepatitis viruses)
- Of chronics: ~20% develop cirrhosis over 20-30 years
- Cirrhosis → HCC risk (HCV accounts for ~1/3 of US liver cancer cases)
- Unlike HBV: HCV-related HCC occurs almost exclusively in setting of cirrhosis
Treatment - DAA revolution:
- Sofosbuvir (NS5B) + Ledipasvir/Velpatasvir (NS5A) ± Glecaprevir/Pibrentasvir (NS3)
- >95% SVR (cure) rates in 8-12 weeks; pan-genotypic regimens available
SECTION 5: HEPATITIS D VIRUS (HDV)
5.1 Microbiology
Exam Trigger: "Defective/satellite virus," "Requires HBsAg," "Delta antigen," "Worst acute hepatitis"
- Unique: HDV is a defective, satellite virus - it can only replicate in cells ALSO infected with HBV, because it BORROWS HBsAg to form its envelope
- Genome: 1.7 kb circular, negative-sense ssRNA - smallest human viral genome
- Inner core: HDV RNA + Hepatitis D antigen (HDAg)
- Outer coat: Borrowed HBsAg from HBV
HDAg - Two Forms:
| Form | Size | Function |
|---|---|---|
| Small HDAg (p24) | 195 aa | Promotes HDAg and HDV RNA replication |
| Large HDAg (p27) | 214 aa | Inhibits replication; required for virion assembly (binds HBsAg) |
5.2 Two Clinical Patterns - EXAM ESSENTIAL
| Co-infection | Superinfection | |
|---|---|---|
| Definition | Simultaneous acquisition of HBV + HDV | HDV infects a CHRONIC HBV CARRIER |
| Serology | IgM anti-HBc + IgM anti-HDV | HBsAg + anti-HDV; IgM anti-HBc NEGATIVE (no new HBV) |
| Outcome | Usually self-limited; both viruses cleared | >80% chronic HDV; severe chronic hepatitis |
| Fulminant risk | Increased (especially IV drug users) | Very high |
| Chronicity | ~10% | >80% |
Serology summary:
- Acute HDV (co-infection): Anti-HDAg develops late, may be low titer; IgM anti-HDV detectable
- HDV superinfection: High, persistent IgM and IgG anti-HD; HBsAg positive; HDV RNA + HDAg persist
- All HDV markers disappear in recovered co-infection
- Anti-HBc IgM differentiates: POSITIVE in co-infection; NEGATIVE in superinfection
PREVENTION PEARL: HBV vaccination completely prevents HDV infection (no HBV = no HDV)
SECTION 6: HEPATITIS E VIRUS (HEV)
6.1 Microbiology
Exam Trigger: "20% mortality in pregnancy," "Zoonosis," "Waterborne epidemic," "Fecal-oral like HAV"
- Classification: Hepeviridae family, genus Hepevirus
- Structure: 30-32 nm, non-enveloped, icosahedral, positive-sense ssRNA (7.2 kb)
- Four genotypes:
- Genotypes 1 & 2: Human only; cause large waterborne epidemics
- Genotypes 3 & 4: Zoonotic (pigs, deer, rabbits, wild boar); cause sporadic cases in developed countries
- Animal reservoirs: Pigs, monkeys, cats, dogs, deer
6.2 Epidemiology
- Endemic regions: Asia (India - HEV = 30-60% of sporadic acute hepatitis!), Indian subcontinent, sub-Saharan Africa, Mexico, Central America
- Sporadic cases: Europe (pig farming), North America (travelers, pork consumption)
- Fecal-oral route; large waterborne outbreaks in resource-poor settings
- Incubation: 4-5 weeks (similar to HAV)
6.3 Immunology
- Self-limiting in immunocompetent hosts - no chronic disease
- Chronic HEV in immunocompromised patients (transplant recipients, HIV): genotype 3 responsible
- Pregnant women - HIGH YIELD:
- Mortality approaches 20% (especially 3rd trimester)
- Mechanism: estrogen/progesterone alter T regulatory cell function and innate immunity → impaired viral clearance → fulminant hepatitis
- Fulminant hepatic failure risk is unique to pregnancy (contrast with HAV, where pregnancy is not a major risk factor)
- Diagnosis: IgM and IgG anti-HEV + PCR for HEV RNA
- Treatment: Ribavirin for chronic/severe cases; no approved vaccine globally (one approved in China only)
SECTION 7: IMMUNOLOGICAL MECHANISMS - COMPARATIVE SUMMARY
7.1 Mechanism of Liver Damage in All Viral Hepatitides
Universal Exam Concept: NONE of the five classical hepatitis viruses are directly cytopathic (or only minimally so). Liver damage = immune-mediated.
INFECTED HEPATOCYTE
↓
Presents viral peptides
on MHC class I
↓
CD8+ Cytotoxic T Lymphocytes (CTLs)
recognize and KILL hepatocyte
↓
Hepatocyte necrosis → ↑ALT/AST
↓
CD4+ T helper cells → IFN-γ, TNF-α
↓
Macrophage activation → more inflammation
↓
B cells → antiviral antibodies (some protective)
7.2 Why Different Viruses Lead to Different Outcomes
| Factor | HAV | HBV | HCV |
|---|---|---|---|
| Immune response strength | Strong, rapid | Strong in adults | Weak, suboptimal |
| T cell exhaustion | No | Yes (chronic) | Major factor |
| Viral immune evasion | Minimal | cccDNA, HBeAg tolerance | NS3/4A cleaves MAVS/TRIF; quasispecies |
| Viral integration | No | Yes (promotes HCC) | No |
| Neutralizing antibody | IgG anti-HAV (protective) | Anti-HBs (protective) | None effective |
7.3 Innate Immunity
- Interferons (IFN-α, IFN-β): First line of defense; upregulate ISGs (interferon-stimulated genes) in hepatocytes; limit early replication
- NK cells: Kill infected cells without prior sensitization; produce IFN-γ
- Pattern recognition: RIG-I and MDA5 detect viral RNA → IRF3/7 → IFN-β production
- HCV specifically BLOCKS this: NS3/4A cleaves MAVS (RIG-I adaptor) and TRIF (TLR3 adaptor) → no IFN-β
7.4 Adaptive Immunity
- CD4+ T cells: Coordinate response; produce IFN-γ (activates macrophages), IL-2 (expands CTLs), IL-4/IL-5 (helps B cells)
- CD8+ CTLs: Primary killers of infected hepatocytes via perforin/granzyme pathway and Fas-FasL
- B cells/Antibodies: Most important for HAV and HBV; anti-HBs is the ONLY protective antibody in hepatitis; anti-HCV is NOT protective
SECTION 8: HISTOPATHOLOGY OF VIRAL HEPATITIS (EXAM POINTS)
8.1 Acute Viral Hepatitis (All Types)
- Hepatocyte swelling (ballooning degeneration)
- Acidophil/Councilman bodies - apoptotic hepatocytes (eosinophilic, shrunken)
- Lobular disarray with hepatocyte dropout
- Inflammatory infiltrate - predominantly lymphocytes
- Cholestasis - bile in canaliculi
8.2 Chronic Viral Hepatitis (Grading & Staging)
- Grade = activity (necroinflammation) | Stage = fibrosis extent
- Interface hepatitis (piecemeal necrosis) - lymphocytes erode hepatocytes at portal-parenchymal junction; hallmark of chronic hepatitis
- Bridging necrosis - connects portal tracts or extends to central veins = severe activity
- Portal tract expansion with lymphocytic infiltrate
- Progressive fibrosis → cirrhosis
8.3 Virus-Specific Histological Clues
| Finding | Virus | Significance |
|---|---|---|
| Ground-glass hepatocytes | HBV | Excess HBsAg in ER; pale pink granular cytoplasm on H&E; confirmed by immunostaining |
| Dense portal lymphoid aggregates (lymphoid follicles) | HCV | Characteristic of chronic HCV |
| Macrovesicular steatosis | HCV genotype 3 | Fat accumulation in hepatocytes |
| Bile duct damage | HCV | Mild damage to portal bile ducts |
SECTION 9: LABORATORY DIAGNOSIS - QUICK REFERENCE
| Virus | First-Line Test | Confirmatory | Monitoring |
|---|---|---|---|
| HAV | IgM anti-HAV (ELISA) | Clinical + epidemiology | IgG anti-HAV (immunity) |
| HBV | HBsAg | HBV DNA (PCR); HBeAg; anti-HBc IgM | HBV DNA quantitative |
| HCV | Anti-HCV ELISA | HCV RNA (PCR) | HCV RNA quantitative; genotyping |
| HDV | Anti-HDV ELISA | HDV RNA; HDAg in liver biopsy | HDV RNA |
| HEV | IgM anti-HEV | HEV RNA (PCR) | IgG anti-HEV |
Exam trap: Anti-HCV positive alone does NOT confirm current infection - must confirm with HCV RNA PCR (may be past/resolved infection)
SECTION 10: VACCINES AND PREVENTION
| Virus | Vaccine | Type | Immunogenicity | Coverage |
|---|---|---|---|---|
| HAV | Havrix, Vaqta, Twinrix | Inactivated HAV | Excellent (>95%) | 2 doses |
| HBV | Engerix-B, Recombivax | Recombinant HBsAg | 95% seroprotection; anti-HBs ≥10 IU/L | 3 doses (0,1,6 months) |
| HCV | None | - | - | Prevention: harm reduction |
| HDV | Prevented by HBV vaccine | N/A | N/A | HBV vaccination = full HDV prevention |
| HEV | Hecolin (China only) | Recombinant | Good | Not globally available |
HBV vaccine mechanism: Recombinant HBsAg induces anti-HBs production → neutralizes virus at point of entry. Anti-HBs ≥10 IU/mL = protective.
SECTION 11: HIGH-YIELD EXAM MEMORY AIDS
"Window Period" in HBV - MUST KNOW
Patient had HBV exposure → HBsAg has CLEARED but anti-HBs NOT YET appeared. ONLY IgM anti-HBc is detectable. Window period: anti-HBc IgM is the SOLE marker.
Fulminant Hepatitis by Virus
- Most common overall cause: HBV (but rare)
- Highest risk in pregnancy: HEV (20% mortality)
- Coinfection doubles risk: HDV + HBV
- HAV fulminant: Very rare (0.1%)
"Superinfection is worse than Co-infection" (HDV Rule)
- Co-infection (HDV+HBV simultaneous): usually self-limited
- Superinfection (HDV into chronic HBV): >80% chronic, high fulminant risk
Chronicity Memory Aid:
HAV → 0%
HEV → 0% (but rare in immunocompromised)
HBV → 5-10% adults | 90-95% neonates
HDV → 10% (co-inf) | >80% (superinfection)
HCV → >80% (highest overall)
"Which antibody is protective?"
- Only anti-HBs (hepatitis B) and anti-HAV IgG confer immunity
- Anti-HCV = NOT protective
- Anti-HDV = NOT protective (still need HBsAg presence)
- Anti-HEV IgG = some protection
Sources: Jawetz, Melnick & Adelberg's Medical Microbiology 28e, pp. 517-527 | Robbins & Kumar Basic Pathology, pp. 599-606 | Medical Microbiology 9e, Murray | Tietz Textbook of Laboratory Medicine 7e
Enumerate, describe and discuss the aetiology of upper and lower GI bleeding with detailed explanation in exam pov
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
I now have comprehensive, detailed content from multiple authoritative surgical and gastroenterological textbooks. Let me compile the complete, exam-focused answer.
🎓 Aetiology of Upper and Lower GI Bleeding — Complete Exam Guide
PART 1: DEFINITIONS AND ANATOMICAL LANDMARK
The Ligament of Treitz (Suspensory ligament of duodenum) is the DIVIDING LINE
| Type | Location | Landmark |
|---|---|---|
| Upper GI Bleeding (UGIB) | Proximal to Ligament of Treitz | Oesophagus, stomach, duodenum |
| Lower GI Bleeding (LGIB) | Distal to Ligament of Treitz | Small bowel, colon, rectum, anus |
Practical Definition of "Massive" UGIB: Acute GI bleeding proximal to the ligament of Treitz requiring blood transfusion.
PART 2: CLINICAL PRESENTATION TERMINOLOGY (EXAM MUST-KNOW)
| Term | Definition | Typical Source |
|---|---|---|
| Haematemesis | Vomiting of blood (bright red or "coffee-ground") | UGIB (oesophagus, stomach, duodenum) |
| Coffee-ground vomiting | Partially digested blood = dark, grainy vomit | UGIB (slower bleeding) |
| Melaena | Black, tarry, foul-smelling stool (digested blood) | UGIB (or proximal small bowel) |
| Haematochezia | Bright red blood per rectum | LGIB; but can be brisk UGIB (15% of cases) |
| Occult bleeding | Positive faecal occult blood test, no visible blood | Any site; often tumours |
Exam Trap: Haematochezia does NOT always mean LGIB. Severe, brisk UGIB causes haematochezia in ~15% of cases. Always exclude upper source first if haemodynamically unstable!
Biochemical Clue: Digested blood is a protein load → raises BUN (blood urea nitrogen) → elevated BUN:Creatinine ratio (>20:1) is a clue to UGIB.
PART 3: CLASSIFICATION FRAMEWORK
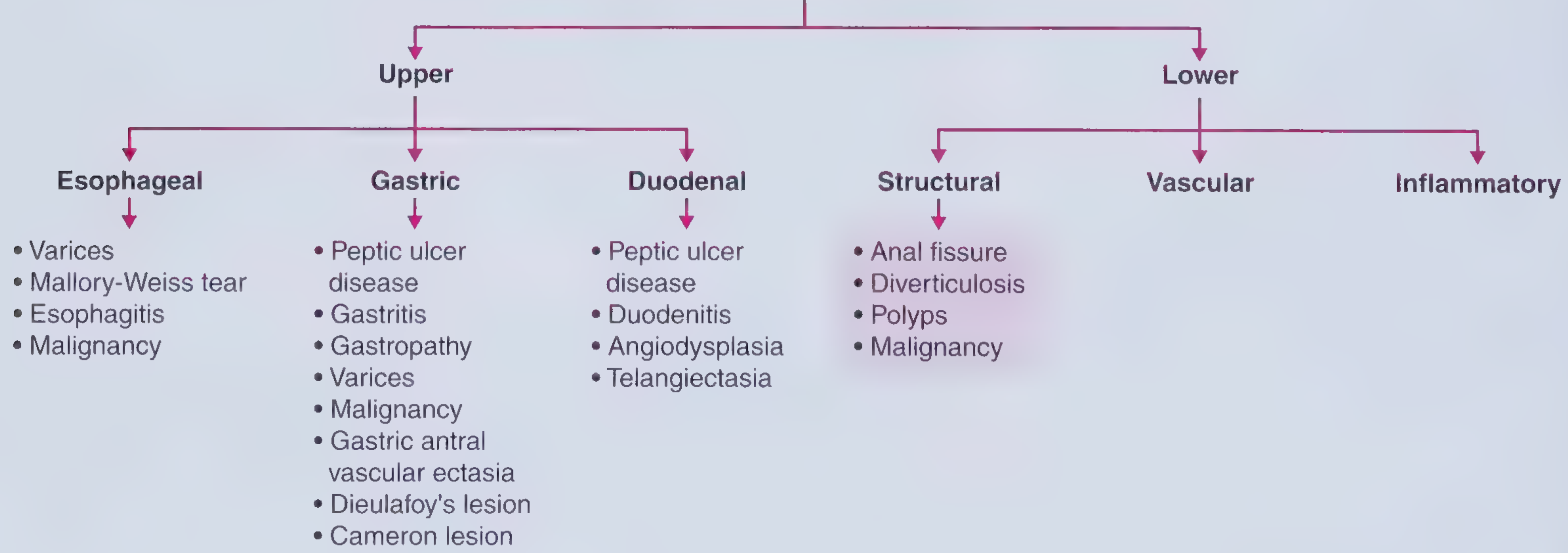
SECTION A: UPPER GASTROINTESTINAL BLEEDING (UGIB)
Epidemiology and Frequency Data (Direct Exam Numbers)
- Incidence: ~50 cases per 100,000 persons per year
- Mortality: 5-10% (unchanged since 1970s despite advances)
- 80% of UGIB is self-limited without specific therapy
- Of the 20% that rebleed: mortality is 30-40%
- Endoscopy identifies the source in 90% of cases
Frequency of causes from UCLA CURE Database (968 cases):
| Cause | Frequency |
|---|---|
| Peptic ulcer | 35-40% (MOST COMMON) |
| Oesophageal/gastric varices | 21.9% |
| Portal hypertension-related lesions | 4.6% |
| Oesophagitis | 4.6% |
| Angioectasia/telangiectasia | 4.0% |
| Mallory-Weiss tear | 4.0% |
| Dieulafoy lesion | 3.2% |
| UGI neoplasm | 3.1% |
| Epistaxis (swallowed blood) | 2.2% |
| Erosions | 1.2% |
| No cause found | 7.3% |
(Sleisenger & Fordtran's Gastrointestinal and Liver Disease; Schwartz's Principles of Surgery 11e)
A1: OESOPHAGEAL CAUSES
1. Oesophageal Varices ⭐⭐⭐ (Most Dangerous Oesophageal Cause)
Definition: Dilated, tortuous submucosal veins in the oesophagus, present in up to 50% of cirrhotics at diagnosis.
Pathogenesis:
Portal hypertension (cirrhosis most common cause)
↓
Increased portal venous pressure
↓
Blood diverted to portosystemic collaterals
↓
Oesophageal veins dilate → VARICES form
↓
Rupture when pressure too high (>12 mmHg threshold)
↓
MASSIVE haematemesis
Aetiology of portal hypertension leading to varices:
- Pre-hepatic: Portal vein thrombosis, splenic vein thrombosis
- Intrahepatic (most common): Cirrhosis (alcohol, HBV, HCV, NASH), schistosomiasis
- Post-hepatic: Budd-Chiari syndrome, right heart failure, constrictive pericarditis
Clinical features:
- Massive, sudden haematemesis in a known cirrhotic
- Associated signs of liver disease: jaundice, spider naevi, palmar erythema, splenomegaly
- Life-threatening - mortality 15-20% per variceal bleed episode; 30% die in 6 weeks if untreated
Key Exam Points:
- Variceal bleeding accounts for the VAST MAJORITY of GI bleeding in cirrhotics
- Gastric varices also occur: Type 1 (fundic), Type 2 (distal - more dangerous)
- Treatment: Octreotide (vasoconstrictor) + endoscopic band ligation + prophylactic antibiotics (ceftriaxone)
- If uncontrolled: TIPS (Transjugular Intrahepatic Portosystemic Shunt)
- Primary prophylaxis: Non-selective beta-blockers (propranolol, nadolol)
2. Mallory-Weiss Tear ⭐⭐
Definition: Longitudinal mucosal laceration at the gastro-oesophageal junction or gastric cardia, caused by sudden increase in intra-abdominal/intragastric pressure.
Pathogenesis:
Forceful retching/vomiting (most common)
↓
Sudden rise in intragastric pressure
↓
Mucosal tear at oesophago-gastric junction
↓
Submucosal arterial bleeding
Causes:
- Severe vomiting (alcoholism most common, pregnancy, bulimia)
- Severe coughing, hiccupping, straining
- Blunt abdominal trauma
- Cardiopulmonary resuscitation
Classic History: Haematemesis AFTER an episode of heavy, forceful retching (blood appears with the 2nd or 3rd vomit - first vomit contains no blood)
Key Exam Points:
- Accounts for up to 15% of UGIB
- Most episodes stop SPONTANEOUSLY (>90% self-limited)
- Rebleeding rate: up to 10%
- Treatment: Endoscopic haemostasis (bipolar electrocoagulation, epinephrine injection, clips, band ligation)
- Contrast with Boerhaave syndrome = full-thickness oesophageal perforation (surgical emergency)
3. Oesophagitis ⭐
Definition: Inflammation of the oesophageal mucosa causing mucosal erosion and bleeding.
Causes:
- Gastro-oesophageal reflux disease (GERD) - most common
- Infections: Candida (immunocompromised), CMV, Herpes simplex
- Pill-induced oesophagitis (bisphosphonates, tetracyclines, NSAIDs, iron tablets)
- Eosinophilic oesophagitis
- Radiation oesophagitis
Clinical features:
- Haematemesis; less haemodynamic instability than other UGIB causes
- Prognosis excellent - low rebleeding risk
- Accounts for ~10% of UGIB
4. Oesophageal Cancer ⭐
- Usually presents with progressive dysphagia and weight loss
- GI bleeding is usually occult (iron-deficiency anaemia)
- Overt bleeding uncommon; occurs when tumour erodes into adjacent vascular structures (aorta)
- Risk factors: smoking, alcohol (squamous cell), GERD/Barrett's (adenocarcinoma)
A2: GASTRIC CAUSES
5. Peptic Ulcer Disease (PUD) ⭐⭐⭐ (MOST COMMON CAUSE OF UGIB OVERALL - 35-40%)
Definition: Disruption of the mucosal barrier allowing acid-pepsin digestion of the gastric or duodenal wall.
Pathogenesis:
Imbalance between AGGRESSIVE and DEFENSIVE factors
↓
Aggressive: H. pylori, NSAIDs, acid, pepsin, bile
Defensive: mucus, bicarbonate, prostaglandins, mucosal blood flow
↓
Mucosal ulceration
↓
Erosion into submucosal blood vessel
↓
BLEEDING
Aetiological factors:
| Factor | Mechanism |
|---|---|
| H. pylori infection | Destroys mucosal barrier; urease, proteases, VacA toxin; CagA disrupts tight junctions; most common cause globally |
| NSAIDs | Inhibit COX-1 → ↓prostaglandins → ↓mucus + ↓bicarbonate + ↓mucosal blood flow; direct topical injury |
| Corticosteroids | Impair mucosal defence; potentiate NSAID injury |
| Smoking | Impairs mucosal healing; increases acid secretion |
| Stress ulcers (Curling's) | Burns, trauma, sepsis - ischaemia of mucosa |
| Stress ulcers (Cushing's) | Head injury/brain operations - vagal hyperactivity → acid hypersecretion |
| Zollinger-Ellison syndrome | Gastrinoma → hypersecretion of acid → multiple peptic ulcers (multiple endocrine neoplasia type 1 association) |
Most dangerous ulcer: Posterior duodenal ulcer eroding the gastroduodenal artery → massive, life-threatening haemorrhage
Key Exam Points:
- Gastric ulcers on the lesser curvature or posterior wall are most common
- Duodenal ulcers: 90% on the anterior wall of the first part (perforate); posterior wall (bleed)
- H. pylori + NSAIDs are the two strongest independent risk factors
- Duodenal ulcers bleed more than gastric ulcers (proximity to gastroduodenal artery)
- Treatment: IV PPI immediately; endoscopic haemostasis within 24 hours; eradicate H. pylori
6. Gastritis and Gastropathy ⭐
Gastritis: Mucosal injury WITH inflammation
Causes:
- H. pylori (Type B gastritis - most common)
- Autoimmune (Type A - affects fundus/body; pernicious anaemia; anti-parietal cell/anti-intrinsic factor antibodies)
- NSAIDs, alcohol, bile reflux, radiation
- Stress (erosive gastritis) - critically ill patients; burns (Curling's), CNS injury (Cushing's)
- Chemical gastropathy: NSAIDs, alcohol
Bleeding pattern: Usually oozing from multiple superficial erosions; rarely massive
7. Gastric Varices
- Occur with portal hypertension (same mechanism as oesophageal varices)
- Isolated gastric varices (type I = fundic; type II = distal including proximal duodenum) occur without oesophageal varices
- More difficult to treat than oesophageal varices
- Management: Cyanoacrylate (glue) injection preferred over band ligation
8. Gastric Antral Vascular Ectasia (GAVE) - "Watermelon Stomach" ⭐
Definition: Distinctive endoscopic appearance of red stripes/spots in the gastric antrum resembling a watermelon - dilated mucosal and submucosal blood vessels.
Associations:
- Chronic renal failure
- Systemic sclerosis (scleroderma) - most common connective tissue disease association
- Cirrhosis (distinct from portal hypertensive gastropathy)
- Bone marrow transplantation
Clinical features: Usually causes chronic occult bleeding and iron-deficiency anaemia; occasionally overt haematemesis
Key Distinction from Portal Hypertensive Gastropathy:
| Feature | GAVE | PHG |
|---|---|---|
| Location | Antrum | Fundus/body |
| Pattern | Red spots/stripes (watermelon) | Mosaic "snake skin" pattern |
| Portal hypertension | May or may not be present | Always present |
| Treatment | Argon plasma coagulation | Beta-blockers, octreotide |
9. Dieulafoy's Lesion ⭐⭐
Definition: An abnormally large (1-3 mm), tortuous arteriole in the gastric submucosa that does NOT underlie an ulcer but erodes through the overlying mucosa and bleeds massively.
Location: 95% within 6 cm of gastro-oesophageal junction along the lesser curvature (where the submucosal vessels are largest)
Pathogenesis: The large artery lies just beneath a tiny (<3 mm) mucosal defect; pulsatile flow causes intermittent, life-threatening arterial haemorrhage
Key Exam Points:
- Presents as sudden, massive haematemesis with NO prior symptoms (no ulcer history, no portal hypertension)
- Notoriously difficult to find endoscopically, especially when bleeding has stopped
- Common cause of obscure GI bleeding (recurrent bleeding without diagnosis)
- Treatment: Endoscopic haemostasis (epinephrine injection + electrocoagulation) is treatment of choice
- Patient may be young and otherwise healthy
10. Cameron Lesion
Definition: Linear gastric erosions/ulcers within the neck of a hiatal hernia, caused by mechanical trauma of the hernia against the diaphragmatic crus combined with acid exposure.
Clinical features:
- Usually asymptomatic; found incidentally at endoscopy
- Occult bleeding causing iron-deficiency anaemia (more common than overt bleeding)
- Treatment: GERD management + endoscopic therapy if needed
11. Gastric Cancer ⭐
- Usually presents with occult bleeding, weight loss, epigastric pain
- Overt haematemesis or melaena possible with ulcerated tumours
- H. pylori is the main risk factor
- Risk factors: gastric atrophy, intestinal metaplasia, diet (smoked/salty foods), blood group A, smoking
A3: DUODENAL CAUSES
12. Duodenal Peptic Ulcer ⭐⭐⭐
- Same pathogenesis as gastric ulcer (see above)
- Most common cause of UGIB - duodenal ulcers bleed more than gastric ulcers
- Posterior duodenal wall ulcers erode into the gastroduodenal artery = catastrophic haemorrhage
- Classic history: intermittent epigastric pain relieved by food (duodenal pattern), then melaena/haematemesis
13. Duodenitis
- Inflammation of duodenal mucosa without frank ulceration
- Causes: H. pylori, NSAIDs, alcohol, Crohn's disease, celiac disease, radiation
- Usually self-limited bleeding; less severe than ulcer bleeding
14. Duodenal Angiodysplasia / Telangiectasia
- Abnormal, ectatic, dilated tortuous small (<10 mm) blood vessels in mucosa/submucosa
- Most common vascular malformation of GI tract
- Typically affects patients >60 years old
- Heyde's syndrome: Association of aortic stenosis + angiodysplasia (platelet dysfunction from shear stress through stenotic valve destroys vWF multimers)
- Associated with: chronic renal failure, radiation, connective tissue disease, hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu)
- Can cause occult or overt bleeding; often intermittent
15. Aorto-enteric Fistula ⭐ (EXAM FAVOURITE - Must Know)
Definition: Abnormal communication between the aorta and the duodenum (most common GI site).
Types:
- Primary: Aortic aneurysm eroding into the duodenum
- Secondary: Aortic graft eroding into the duodenum (after aortic reconstruction surgery)
Classic presentation:
- "Herald bleed" - initial minor, self-limiting haematemesis/melaena
- Asymptomatic interval (hours to days)
- Catastrophic, exsanguinating haemorrhage - fatal if not immediately recognised and operated
Key Exam Point: In ANY patient with haematemesis/melaena + history of aortic graft/aneurysm repair, ALWAYS consider aorto-enteric fistula first - it is a surgical emergency.
SECTION B: LOWER GASTROINTESTINAL BLEEDING (LGIB)
Epidemiology
- Incidence: ~20 cases per 100,000 per year (less than UGIB)
- Most patients >70 years of age
- Presents with painless haematochezia
- Majority (80-90%) stop spontaneously
- If haematochezia + orthostasis: exclude brisk UGIB first (15% of haematochezia from UGIB)
Frequency of LGIB causes:
| Cause | Frequency |
|---|---|
| Diverticulosis | ~30% (MOST COMMON) |
| Colonic polyps / cancer | ~20% |
| Colitis (IBD, infectious, ischaemic) | ~20% |
| Anorectal disorders (haemorrhoids, fissure) | ~20% |
| Angiodysplasia | Common in elderly |
| Small bowel source | 5% |
(Sleisenger & Fordtran's GI and Liver Disease)
B1: STRUCTURAL CAUSES OF LGIB
1. Diverticular Disease ⭐⭐⭐ (MOST COMMON CAUSE OF LGIB)
Definition: Acquired outpouchings of the colonic mucosa/submucosa through weak points in the muscular wall (at sites where blood vessels penetrate).
Pathogenesis of bleeding:
Diverticulum forms at weak point in colonic wall
↓
Vasa recta (penetrating blood vessel) is stretched
over the dome of the diverticulum
↓
Intimal thickening + medial thinning
↓
Vessel ruptures into diverticulum lumen
↓
MASSIVE painless haematochezia
Epidemiology:
- Prevalence increases with age: 5% at age 40 → >65% by age 80
- Western diet (low fibre) major risk factor
- Right-sided diverticula bleed more (despite left-sided being more common)
Key Exam Points:
- Most common cause of LGIB requiring hospitalisation
- Bleeding is typically sudden, PAINLESS, massive haematochezia (dark red or maroon)
- No abdominal pain differentiates it from diverticulitis (which causes LEFT iliac fossa pain and fever)
- 75-80% stop spontaneously
- Rebleeding rate: 25-30% within 4 years
- Treatment: Endoscopic haemostasis; selective angioembolisation; surgery if uncontrolled
2. Colorectal Polyps ⭐
Types:
- Neoplastic (adenomatous): Tubular, tubulovillous, villous adenomas - premalignant
- Hamartomatous: Peutz-Jeghers syndrome (pigmented lips), juvenile polyps
- Inflammatory: Post-inflammatory (pseudopolyps in IBD)
- Hyperplastic: Usually non-neoplastic
Bleeding characteristics:
- Usually occult bleeding causing iron-deficiency anaemia
- Overt haematochezia can occur, especially from rectal polyps
- Villous adenomas of the rectum can cause significant rectal bleeding + mucous diarrhoea + hypokalaemia
- Postpolypectomy bleeding accounts for ~10% of LGIB
3. Colorectal Cancer ⭐⭐
Bleeding pattern:
- Left-sided/rectal cancer: Overt bright red haematochezia (closer to anus, less time for blood to change)
- Right-sided/caecal cancer: Occult bleeding → iron-deficiency anaemia, fatigue (blood mixes with stool)
- Change in bowel habit + iron-deficiency anaemia in >50-year-old = colorectal cancer until proved otherwise
Key Exam Features of Right-Sided vs Left-Sided:
| Feature | Right-sided (caecum/ascending) | Left-sided (descending/sigmoid/rectum) |
|---|---|---|
| Presentation | Occult bleeding, anaemia, mass | Overt haematochezia, altered bowel habit, obstruction |
| Stool | Normal (may have IDA) | Pencil-thin, ribbon stools, haematochezia |
| Obstruction | Rare (wide lumen) | Common (narrow lumen) |
4. Anal Fissure ⭐
Definition: A tear in the mucosa of the anal canal, usually in the posterior midline (90%).
Pathogenesis:
- Hard stool tears the delicate anal canal mucosa
- Hypertonia of internal anal sphincter maintains the fissure (prevents healing)
- Chronic ischaemia due to poor blood supply in the posterior midline
Clinical features:
- Severe pain ON and AFTER defecation (tearing pain)
- Bright red blood ON THE SURFACE of stool or toilet paper (not mixed with stool)
- Sentinel pile (skin tag) at outer end
Key Exam Points:
- Non-midline fissures: suspect Crohn's disease, anal cancer, syphilis, HIV
- First-line: Increased fibre/water, sitz baths, topical glyceryl trinitrate (GTN) or diltiazem
- Refractory: Botulinum toxin injection / lateral internal sphincterotomy
B2: VASCULAR CAUSES OF LGIB
5. Haemorrhoids (Piles) ⭐⭐⭐ (Most Common Anorectal Cause)
Definition: Varicosities/cushions of the anal canal venous plexus.
Classification:
| Type | Location | Venous Plexus | Pain? |
|---|---|---|---|
| Internal | Above dentate line | Superior haemorrhoidal plexus | Painless (above pain-sensitive epithelium) |
| External | Below dentate line | Inferior haemorrhoidal plexus | Painful (below dentate line) |
Grading of Internal Haemorrhoids:
| Grade | Description |
|---|---|
| I | Bleeding without prolapse |
| II | Prolapse on straining; spontaneous reduction |
| III | Prolapse on straining; manual reduction needed |
| IV | Permanently prolapsed; irreducible |
Pathogenesis:
- Low-fibre diet → straining → ↑ intra-abdominal pressure → vascular cushion engorgement
- Pregnancy, chronic constipation, portal hypertension, pelvic tumours
Bleeding pattern:
- Painless bright red blood - seen on the surface of stool, in the toilet pan, or on toilet paper
- Typically at the END of defecation
- Second most common cause of LGIB requiring hospitalisation
Key Exam Points:
- NEVER attribute rectal bleeding to haemorrhoids without excluding cancer
- Internal haemorrhoids are PAINLESS when they bleed
- Pain + bleeding = thrombosed external haemorrhoid or anal fissure
- Treatment: Conservative (fibre, sitz baths) → Rubber band ligation (Grade II-III) → Surgery (haemorrhoidectomy Grade III-IV)
6. Angiodysplasia (Colonic Vascular Ectasia) ⭐⭐
Definition: Acquired, ectatic, dilated, thin-walled small vessels (<10 mm) in the mucosal/submucosal layers of the GI tract.
Pathogenesis (Laplace's Law mechanism):
Ageing → degenerative changes in submucosal veins
↓
Repeated episodes of raised intraluminal pressure
during muscular contractions
↓
Vascular dilation (Laplace's law: T = P × r, larger radius = more tension)
↓
Arteriovenous communications develop
↓
Bleeding from thin-walled vessels
Key Locations: Caecum and right colon (most common); also small bowel
Associations:
- Age >60 (most important risk factor)
- Chronic renal failure (most common in dialysis patients)
- Aortic stenosis (Heyde's syndrome - acquired vWF deficiency)
- Hereditary Haemorrhagic Telangiectasia (Osler-Weber-Rendu)
- Von Willebrand disease
Key Exam Points:
- Can cause occult, intermittent overt, or massive haematochezia
- Cannot be seen on barium enema - requires colonoscopy or angiography
- Treatment: Endoscopic argon plasma coagulation (APC); angioembolisation; surgery
7. Telangiectasias
- Associated with systemic sclerosis (scleroderma): CREST syndrome (Calcinosis, Raynaud's, Oesophageal dysmotility, Sclerodactyly, Telangiectasia)
- Hereditary Haemorrhagic Telangiectasia (HHT/Osler-Weber-Rendu): Autosomal dominant; mucocutaneous telangiectasias throughout the GI tract; recurrent GI bleeding
8. Haemangiopericytoma / Haemangioma
- Benign vascular tumours; can bleed; usually in small bowel; diagnosed on CT angiography or capsule endoscopy
9. Kaposi Sarcoma
- Malignant vascular tumour; seen in AIDS patients (CD4 <200); also in immunosuppressed transplant patients
- Violaceous endoscopic lesions throughout the GI tract
B3: INFLAMMATORY CAUSES OF LGIB
10. Inflammatory Bowel Disease (IBD) ⭐⭐
Ulcerative Colitis (UC) - More commonly causes haematochezia:
| Feature | Ulcerative Colitis | Crohn's Disease |
|---|---|---|
| Location | Rectum → colon (continuous) | Anywhere mouth to anus (skip lesions) |
| Bleeding | Bloody diarrhoea - hallmark | Less common; usually occult |
| Depth | Mucosa/submucosa only | Transmural (full thickness) |
| GI bleed type | Haematochezia with mucus diarrhoea | Variable; may have perianal disease |
Pathogenesis of bleeding in IBD:
- Mucosal ulceration → erosion of submucosal vessels
- Deep ulcers in Crohn's can erode major vessels → massive haemorrhage
Key Exam Points:
- UC: Bloody mucoid diarrhoea is the cardinal symptom
- UC is associated with: pyoderma gangrenosum, erythema nodosum, primary sclerosing cholangitis, ankylosing spondylitis
- Massive haemorrhage in UC requiring emergency colectomy = <2% of cases
11. Ischaemic Colitis ⭐⭐
Definition: Inadequate blood flow to the colon causing mucosal ischaemia and haemorrhage.
Aetiology:
- Hypotension/shock (most common precipitant) - non-occlusive ischaemia
- Atherosclerosis of mesenteric arteries
- Vasculitis
- Colon surgery (ligation of inferior mesenteric artery)
- Hypercoagulable states, cocaine use, oral contraceptive pill
Most vulnerable sites:
- Watershed areas = splenic flexure (between SMA and IMA territories) and sigmoid colon (rectosigmoid junction)
Clinical features:
- Sudden onset crampy left-sided abdominal pain followed by haematochezia - classic triad
- Moderate, self-limited bleeding (not usually massive)
- Can progress to gangrenous ischaemia → peritonitis, perforation
X-ray sign: "Thumbprinting" = submucosal oedema/haemorrhage creating scalloped indentations on the colonic wall
Exam Mnemonic: Ischaemic colitis = "Splenic flexure + pain + haematochezia + precipitating event (shock/surgery)"
12. Infectious Colitis ⭐
Causative organisms:
| Organism | Feature |
|---|---|
| Campylobacter jejuni | Most common bacterial cause; poultry-related; painful bloody diarrhoea |
| E. coli O157:H7 (EHEC) | Haemorrhagic colitis; may cause HUS (haemolytic uraemic syndrome) = bloody diarrhoea + haemolytic anaemia + thrombocytopenia + acute kidney injury |
| Salmonella, Shigella | Dysentery; bloody mucoid diarrhoea |
| Clostridioides difficile | Post-antibiotic; pseudomembranous colitis; can cause bloody diarrhoea |
| Entamoeba histolytica | Amoebic dysentery; "flask-shaped" ulcers; liver abscess risk |
| CMV colitis | In immunocompromised (HIV, transplant) |
Exam Alert: "Dozens of patrons of a fast-food restaurant develop bloody diarrhoea" = E. coli O157:H7 / Campylobacter outbreak. "Bloody diarrhoea + haemolytic anaemia + thrombocytopenia + AKI" = HUS from EHEC.
13. Radiation Coloproctitis ⭐
- Delayed complication of pelvic radiotherapy (prostate, cervical, rectal cancer)
- Endarteritis of submucosal vessels → mucosal ischaemia + telangiectasia formation
- Chronic, recurrent haematochezia months to years after radiation
- Endoscopy: friable, telangiectatic rectal/colonic mucosa
- Treatment: Argon plasma coagulation (APC) - first-line endoscopic therapy; sucralfate enemas; hyperbaric oxygen
SECTION C: SMALL BOWEL (OBSCURE GI BLEEDING) (Exam Bonus)
Sources of bleeding from the small bowel (between ligament of Treitz and ileocaecal valve) account for 5% of GI bleeding and are the primary cause of "obscure GI bleeding" (bleeding that persists/recurs after negative upper and lower endoscopy).
Causes:
- Angiodysplasia (most common)
- Small bowel tumours (GIST, carcinoid, lymphoma, adenocarcinoma)
- Crohn's disease
- Meckel's diverticulum (ectopic gastric mucosa → peptic ulceration)
- NSAIDs-induced small bowel ulcers
- Coeliac disease
- Small bowel varices (portal hypertension)
Diagnostic tools: Capsule endoscopy (first-line non-invasive); double-balloon enteroscopy; CT enterography; nuclear scintigraphy (Meckel's scan with Tc-99m pertechnetate)
SECTION D: SUMMARY COMPARISON TABLE (Exam Quick Reference)
| Feature | UGIB | LGIB |
|---|---|---|
| Landmark | Proximal to Ligament of Treitz | Distal to Ligament of Treitz |
| Presentation | Haematemesis, melaena, coffee-ground vomiting | Haematochezia (usually), occult bleeding |
| BUN:Creatinine ratio | Elevated (>20:1) | Normal |
| NGT aspirate | Blood or coffee-grounds | Usually clear (but bilious clear does not exclude duodenal ulcer) |
| Incidence | ~50/100,000/yr | ~20/100,000/yr |
| Most common cause | Peptic ulcer disease (35-40%) | Diverticulosis (~30%) |
| Most dangerous | Variceal bleeding (30% 6-week mortality) | Aorto-enteric fistula (if from missed UGIB) |
| Spontaneous cessation | 80% | 80-90% |
| Investigation of choice | OGD within 24 hours | Colonoscopy within 24 hours |
| Mortality | 5-10% | Lower; depends on cause |
SECTION E: RISK STRATIFICATION SCORES (Clinical Exam Points)
Rockall Score (UGIB) - Predicts Rebleeding and Mortality
| Variable | Score |
|---|---|
| Age <60 / 60-79 / ≥80 | 0 / 1 / 2 |
| No shock / Pulse >100 / BP <100 | 0 / 1 / 2 |
| No comorbidity / Cardiac failure/IHD / Renal/liver/metastases | 0 / 2 / 3 |
| Post-endoscopy: Mallory-Weiss/no lesion | 0 |
| Post-endoscopy: ulcer, erosions, oesophagitis | 1 |
| Post-endoscopy: malignancy, varices | 2 |
| Forrest classification (endoscopic stigmata) | |
| Forrest Ia: Active arterial spurting | High risk rebleed |
| Forrest Ib: Active ooze | High risk |
| Forrest IIa: Non-bleeding visible vessel | High risk (50% rebleed) |
| Forrest IIb: Adherent clot | Intermediate risk |
| Forrest IIc: Flat pigmented spot | Low risk |
| Forrest III: Clean base | Low risk (<5% rebleed) |
Glasgow-Blatchford Score (GBS) - Pre-endoscopy; predicts need for intervention
- Uses: BUN, haemoglobin, systolic BP, heart rate, melena, syncope, liver disease, cardiac failure
- Score 0 = safe to discharge without urgent endoscopy
SECTION F: HIGH-YIELD EXAM MNEMONICS
Causes of UGIB - "PEP + M-D-G-A"
- P - Peptic ulcer (most common)
- E - Esophageal/gastric varices
- P - Portal hypertensive gastropathy
- M - Mallory-Weiss tear
- D - Dieulafoy's lesion
- G - GAVE (watermelon stomach)
- A - Aorto-enteric fistula (must never miss)
Causes of LGIB - "DIVERT-CAHIR"
- D - Diverticulosis (most common)
- I - IBD (Crohn's + UC)
- V - Vascular (angiodysplasia, haemorrhoids, telangiectasia)
- E - Ectasia (GAVE)
- R - Rectal cancer/polyps
- T - Trauma/radiation proctitis
- C - Colitis (infectious, ischaemic)
- A - Anal fissure
- H - Haemorrhoids
- I - Ischaemic colitis
- R - Radiation coloproctitis
"Posterior ulcer Bleeds, Anterior ulcer Perforates" (DU locations)
Sources: Frameworks for Internal Medicine (pp. 218-225) | Sleisenger & Fordtran's GI and Liver Disease (pp. 304-320) | Schwartz's Principles of Surgery 11e (pp. 1180-1185) | Maingot's Abdominal Operations | Fischer's Mastery of Surgery 8e | Tintinalli's Emergency Medicine
This is a shared conversation. Sign in to Orris to start your own chat.