Pyrimidine catabolism
Pyrimidine Catabolism
Key Contrast with Purine Catabolism
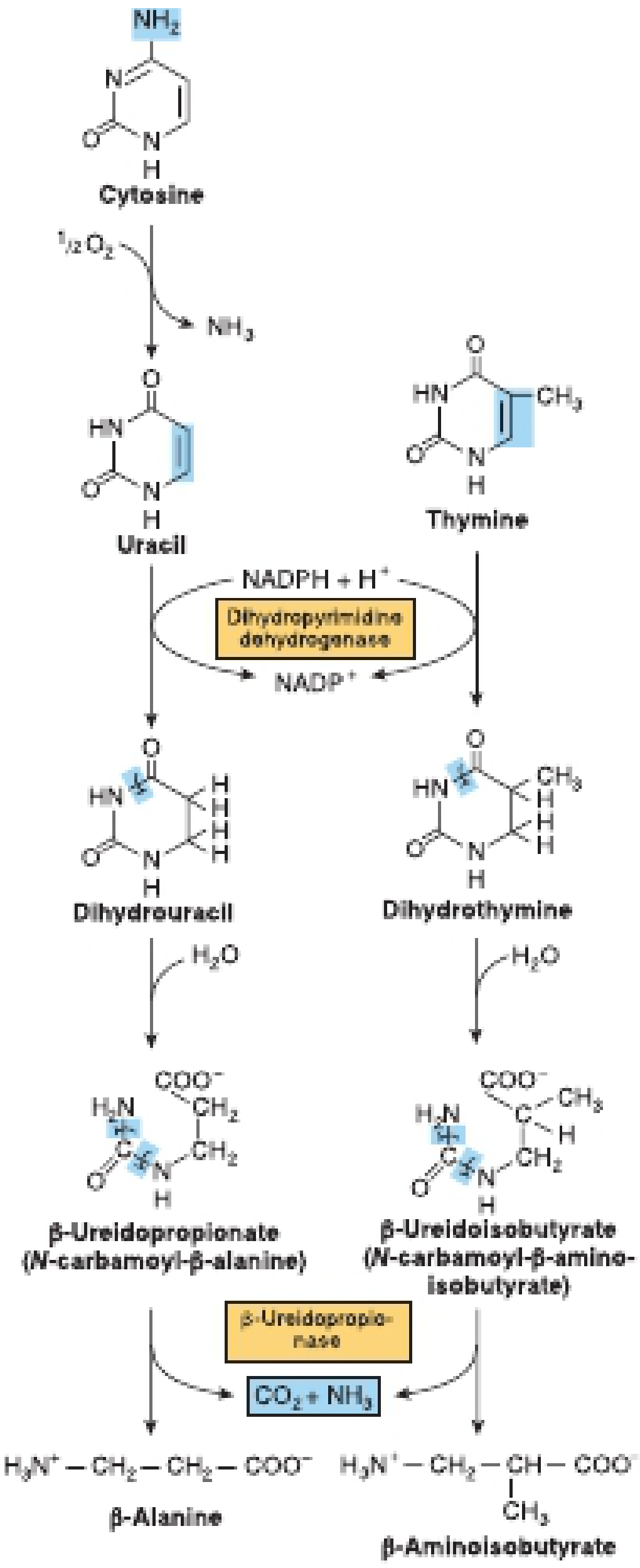
The Three-Step Pathway
Step 1 - Reduction (Ring Saturation)
- Uracil → Dihydrouracil
- Thymine → Dihydrothymine
Step 2 - Ring Opening (Hydrolysis)
- Dihydrouracil + H2O → β-Ureidopropionate (N-carbamoyl-β-alanine)
- Dihydrothymine + H2O → β-Ureidoisobutyrate (N-carbamoyl-β-aminoisobutyrate)
Step 3 - Final Hydrolysis
- β-Ureidopropionate → β-Alanine + CO2 + NH3
- β-Ureidoisobutyrate → β-Aminoisobutyrate + CO2 + NH3
Fate of End Products
| End Product | Source | Fate |
|---|---|---|
| β-Alanine | Uracil (and cytosine via uracil) | Excreted in urine; or transaminated to malonate semialdehyde → acetyl-CoA |
| β-Aminoisobutyrate | Thymine | Excreted in urine; or converted to succinyl-CoA (enters TCA cycle) |
| CO2 | Both pathways | Exhaled |
| NH3 | Both pathways | Enters urea cycle |
Pseudouridine - The Exception
Clinical Disorders of Pyrimidine Catabolism
1. Dihydropyrimidine Dehydrogenase (DPD) Deficiency
- Also called β-hydroxybutyric aciduria or combined uraciluria-thyminuria
- Genetic form: autosomal recessive; causes serious neurological complications (developmental delay, autistic features, seizures - as described in Bradley and Daroff's Neurology in Clinical Practice)
- Because DPD is blocked, both uracil and thymine accumulate and are excreted in urine
- This is simultaneously a disorder of β-amino acid biosynthesis, since β-alanine and β-aminoisobutyrate cannot be formed
- Critical pharmacological implication: 5-fluorouracil (5-FU), the anticancer drug, is a fluorinated uracil analog that is normally detoxified by DPD. Patients with low DPD levels (genetic or otherwise) who receive 5-FU suffer severe, potentially fatal 5-FU toxicity (mucositis, myelosuppression, neurotoxicity)
2. Dihydropyrimidinase Deficiency
- Second enzyme in the pathway (ring opening step)
- Also associated with severe 5-FU toxicity in cancer patients
- Neurological complications including autistic features
3. β-Ureidopropionase Deficiency
- Third enzyme; impairs the final hydrolysis step
- Similarly associated with severe 5-FU toxicity
Summary Table (from Harper's Table 33-1)
| Defective Enzyme | Key Signs |
|---|---|
| Dihydropyrimidine dehydrogenase | β-hydroxybutyric aciduria, uraciluria-thyminuria, neurological disease, severe 5-FU toxicity |
| Dihydropyrimidinase | Neurological disease, severe 5-FU toxicity |
| β-Ureidopropionase | β-amino acid deficiency, severe 5-FU toxicity |
Overproduction Scenarios
- Hyperuricemia with PRPP overproduction - excess PRPP drives both purine AND pyrimidine synthesis, increasing β-alanine excretion
- Ornithine transcarbamoylase (OTC) deficiency - excess carbamoyl phosphate exits mitochondria into the cytosol and drives pyrimidine biosynthesis, causing orotic aciduria + uraciluria + uridinuria
- Allopurinol / 6-azauridine - both drugs can precipitate orotic aciduria by inhibiting orotidylate decarboxylase in pyrimidine biosynthesis
- Reye syndrome - damaged mitochondria cannot use carbamoyl phosphate, which spills into the cytosol and drives orotic acid overproduction
- Harper's Illustrated Biochemistry, 32nd Ed., pp. 349, 357-358 (Figure 33-12)
- Basic Medical Biochemistry: A Clinical Approach, 6th Ed., Chapter 39
- Bradley and Daroff's Neurology in Clinical Practice - dihydropyrimidine dehydrogenase deficiency section
Pyrimidine degradation flowchart photo
Pyrimidine degradation flowchart showing: Cytosine being deaminated to Uracil (releasing NH3), and Thymine on a parallel track. Step 1: Both Uracil and Thymine reduced by Dihydropyrimidine dehydrogenase (DPD) using NADPH to form Dihydrouracil and Dihydrothymine respectively. Step 2: Ring opening by Dihydropyrimidinase with H2O to form beta-Ureidopropionate (N-carbamoyl-beta-alanine) from Dihydrouracil, and beta-Ureidoisobutyrate (N-carbamoyl-beta-aminoisobutyrate) from Dihydrothymine. Step 3: Final hydrolysis by beta-Ureidopropionase releasing CO2 and NH3, producing beta-Alanine from the uracil pathway and beta-Aminoisobutyrate from the thymine pathway. Clean educational biochemistry diagram with two parallel columns, arrows, enzyme names in boxes, cofactors labeled, and final products highlighted.
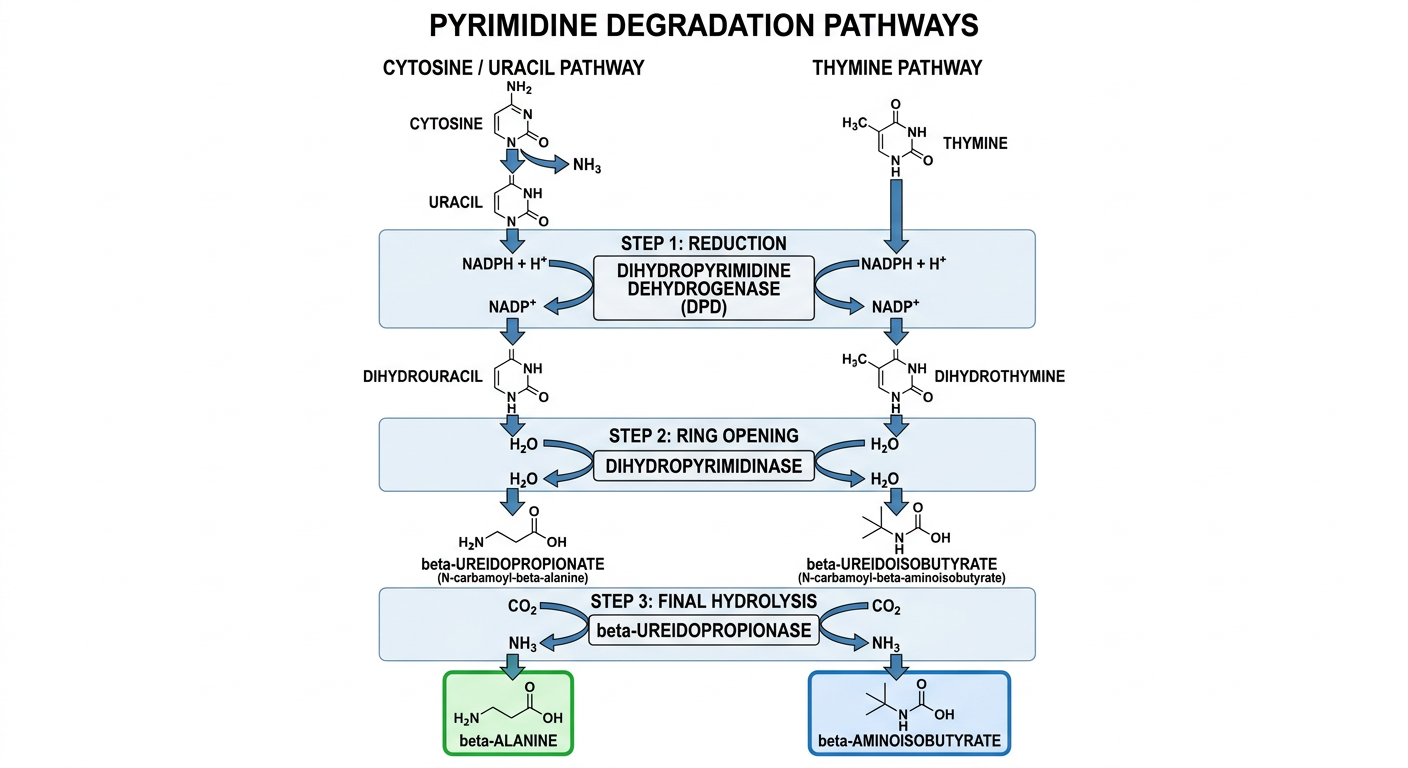
| Step | Uracil Track | Thymine Track |
|---|---|---|
| 1 - Reduction | Uracil + NADPH → Dihydrouracil | Thymine + NADPH → Dihydrothymine |
| Enzyme | Dihydropyrimidine dehydrogenase (DPD) | Same enzyme |
| 2 - Ring opening | Dihydrouracil + H2O → β-Ureidopropionate | Dihydrothymine + H2O → β-Ureidoisobutyrate |
| Enzyme | Dihydropyrimidinase | Same enzyme |
| 3 - Final hydrolysis | β-Ureidopropionate → β-Alanine + CO2 + NH3 | β-Ureidoisobutyrate → β-Aminoisobutyrate + CO2 + NH3 |
| Enzyme | β-Ureidopropionase | Same enzyme |