Aml in detail
Reading File
Finding Sources
Searching PubMed
"acute myeloid leukemia"[MeSH Terms] AND "therapy"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Here is a detailed, textbook-based overview of Acute Myeloid Leukemia (AML):
Acute Myeloid Leukemia (AML) - In Detail
Definition
AML is a tumor of hematopoietic progenitors caused by acquired oncogenic mutations that impede differentiation, leading to the accumulation of immature myeloid blasts in the bone marrow and peripheral blood. The replacement of normal marrow with blasts produces marrow failure and complications related to anemia, thrombocytopenia, and neutropenia. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Epidemiology
- Most common acute leukemia in adults; median age at diagnosis is 60 years
- Incidence rises with age: ~10/100,000/year in those over 60
- ~13,000 new cases per year in the United States
- In children, AML accounts for approximately one-third of acute leukemias; ALL dominates in this age group
- Risk factors: ionizing radiation, benzene, cytotoxic chemotherapy, smoking; most cases are idiopathic
Pathogenesis
Driver mutations fall into four functional categories (Robbins, Cotran & Kumar):
1. Transcription Factor Mutations
- t(8;21) - disrupts RUNX1 (core binding factor-alpha), blocks myeloid maturation
- inv(16) - disrupts CBFB (core binding factor-beta)
- Both create fusion proteins that interfere with the RUNX1/CBFB transcription factor complex required for normal hematopoiesis
- t(15;17) (APL) - creates PML-RARα fusion protein that represses transcription of genes needed for myeloid differentiation
2. Signaling Pathway Mutations (proliferation)
- FLT3-ITD (internal tandem duplication) - constitutively activates myeloid cell proliferation; present in ~30% of AML
- KIT mutations (exons 8 and 17) - adversely affect prognosis in CBF-AML (~30%)
- RAS mutations - less common activating mutations
3. Epigenetic Regulators
- DNMT3A, TET2, IDH1/IDH2 mutations - produce the oncometabolite 2-hydroxyglutarate, disrupting DNA methylation and histone modification
- ASXL1, EZH2, SRSF2, SF3B1, U2AF1, ZRSR2, STAG2, BCOR - associated with myelodysplastic features and adverse prognosis
4. NPM1 Mutations
- NPM1 mutations cause abnormal cytoplasmic localization of nucleophosmin; one of the most common recurrent mutations in AML (~30%)
- Favorable prognosis when present without concurrent FLT3-ITD
Classification (WHO 5th Edition, 2022)
The current WHO classification divides AML into two major categories: (Harrison's Principles of Internal Medicine 22E)
I. AML with Specific Genetic Aberrations
| Subtype | Fusion Gene | Prognosis | Key Features |
|---|---|---|---|
| t(8;21)(q22;q22) | RUNX1::RUNX1T1 | Favorable | Full myelocytic maturation, Auer rods, CD19+, slender Auer rods |
| inv(16)(p13.1q22) | CBFB::MYH11 | Favorable | Myelomonocytic differentiation, abnormal eosinophils with basophilic granules |
| t(15;17)(q22;q12) | PML::RARA (APL) | Very Favorable (~85% cured) | Prominent Auer rods in bundles, DIC common, responds to ATRA |
| t(9;11)(p21.3;q23.3) | KMT2A::MLLT3 | Poor | Monocytic differentiation, gingival hypertrophy, DIC risk |
| t(6;9)(p23;q34) | DEK::NUP214 | Very Poor | Basophilia, FLT3-ITD in ~70%, multilineage dysplasia |
| inv(3) / t(3;3) | GATA2, MECOM | Very Poor | Multilineage dysplasia, atypical megakaryocytes, normal/increased platelets |
| t(1;22) | RBM15::MKL1 | Good | Pediatric; Down syndrome-associated, megakaryoblastic |
| NPM1 mutation | - | Favorable (without FLT3-ITD) | "Cup-shaped" blasts on morphology |
| Biallelic CEBPA (bZIP) | - | Favorable | |
| KMT2A rearrangement | Multiple partners | Poor | |
| NUP98 rearrangement | Multiple (cryptic) | Variable | Often cryptic on cytogenetics |
| AML, myelodysplasia-related (AML-MR) | del(5q), del(7q), ASXL1, SRSF2, etc. | Poor | Prior MDS history or MDS-associated mutations |
| Therapy-related AML | - | Very Poor | Prior cytotoxic/radiation exposure |
II. AML Defined by Differentiation (NOS types, no specific genetic aberration)
- AML with minimal differentiation
- AML without maturation
- AML with maturation
- Acute myelomonocytic leukemia
- Acute monocytic leukemia
- Acute erythroid leukemia
- Acute megakaryoblastic leukemia
- Acute basophilic leukemia
Diagnosis
Peripheral Blood & Bone Marrow
- Diagnosis requires ≥20% blasts in the marrow or blood (note: CBF-AML and APL are diagnostic regardless of blast count)
- Myeloblasts: central nuclei, fine chromatin, prominent nucleoli (3-5), variable cytoplasm with possible granules
- Auer rods - eosinophilic rod-like cytoplasmic inclusions; essentially pathognomonic for AML when present; formed from abnormal primary granule fusion
Immunophenotyping (Flow Cytometry)
- Immature AML: CD34, CD117, HLA-DR
- More differentiated: CD13, CD33
- Monocytic AML: CD14, CD15, CD11b, CD64, CD36
- Erythroid: CD36, CD71, CD235a (glycophorin A)
- Megakaryocytic: CD41, CD61
- 10-20% of AML co-expresses lymphoid antigens (CD2, CD7, CD19, CD56) - "aberrant" phenotypes useful for MRD monitoring
Cytochemistry (less used now)
- MPO (myeloperoxidase), Sudan Black B (SBB), CAE (chloroacetate esterase) - positive in myeloid lineage
- NSE (non-specific esterase / ANA, ANB) - positive in monocytic AML; inhibited by sodium fluoride
Molecular & Cytogenetic Workup
- Conventional karyotype (G-banding)
- FISH for t(15;17), t(8;21), inv(16) at diagnosis
- RT-PCR / qRT-PCR for disease monitoring (MRD)
- NGS panels: NPM1, FLT3, CEBPA, IDH1/IDH2, TP53, DNMT3A, ASXL1, RUNX1, SRSF2, U2AF1, and others
Clinical Presentation
Symptoms of marrow failure:
- Fatigue, pallor (anemia)
- Infections, fever, mouth ulcers (neutropenia)
- Bruising, petechiae, bleeding (thrombocytopenia)
Signs of leukemic infiltration:
- Gum hypertrophy (especially monocytic AML)
- Skin nodules/infiltration (leukemia cutis)
- Lymphadenopathy, splenomegaly, hepatomegaly (less prominent than in ALL)
- Back pain / lower extremity weakness (spinal granulocytic/myeloid sarcoma, especially with t[8;21])
- CNS involvement: papilledema, retinal infiltrates, cranial nerve palsies
Lab abnormalities:
- Elevated WBC (may be very high = hyperleukocytosis) or low WBC with circulating blasts
- Elevated LDH, uric acid, phosphorus (tumor lysis risk)
- DIC (especially in APL and monocytic AML)
Prognostic Risk Stratification (ELN 2022 Criteria)
| Risk Group | Genetic Features | Approximate Cure Rate |
|---|---|---|
| Favorable | t(8;21), inv(16), APL/t(15;17), NPM1 mut without FLT3-ITD, biCEBPA | 55-85% |
| Intermediate | CN-AML without favorable/adverse markers, t(9;11), FLT3-ITD with NPM1 mut | ~40% |
| Adverse | TP53 mut, complex karyotype, inv(3), t(6;9), -7, -5/del(5q), ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2 | Very poor without transplant |
Clinical factors that worsen prognosis:
- Older age (most important clinical factor)
- Prior MDS or myeloproliferative neoplasm
- Secondary/therapy-related AML
- High WBC at presentation
- Poor performance status
Treatment
Initial Workup Before Treatment
CBC, chemistry, coagulation studies (PT/PTT/fibrinogen/D-dimer), viral serologies, echo/MUGA, bone marrow aspirate + biopsy with full molecular profiling, HLA typing, central venous access placement.
1. Induction Chemotherapy (Non-APL AML)
The "7+3" regimen remains the standard backbone:
- Cytarabine 100-200 mg/m² as a continuous IV infusion for 7 days (S-phase specific antimetabolite, inhibits DNA synthesis via triphosphate form)
- Anthracycline on days 1, 2, 3: daunorubicin 60-90 mg/m² or idarubicin 12 mg/m² (topoisomerase II inhibitors, DNA intercalators)
- Gemtuzumab ozogamicin (anti-CD33 immunoconjugate) can be added for favorable/intermediate risk patients
Targeted additions based on molecular profile:
- FLT3-mutated AML: add midostaurin to 7+3 (RATIFY trial, 2017) or gilteritinib for relapsed/refractory disease
- IDH1-mutated AML: ivosidenib (approved for newly diagnosed IDH1-mutant AML)
- IDH2-mutated AML: enasidenib
- KMT2A/NPM1-rearranged AML: revumenib (menin inhibitor, 2023)
- Older/unfit patients: venetoclax + azacitidine or decitabine (hypomethylating agent combinations)
2. Post-Remission (Consolidation) Therapy
- Favorable risk (CBF-AML, NPM1 without FLT3-ITD): high-dose cytarabine (HiDAC) consolidation; allogeneic HCT generally not needed in CR1
- Intermediate risk: allogeneic HCT in CR1 if a suitable donor is available
- Adverse risk: allogeneic HCT in CR1 is strongly recommended
3. Acute Promyelocytic Leukemia (APL) - Special Treatment
APL is a hematologic emergency due to high DIC risk. Treatment is unique:
- ATRA (all-trans retinoic acid) + ATO (arsenic trioxide) - standard of care for non-high-risk APL
- ATRA at pharmacologic doses binds to PML-RARα, displaces transcriptional repressors, and restores differentiation gene expression; liganded PML-RARα is also targeted for proteasomal degradation
- The result is terminal differentiation of malignant promyelocytes into short-lived neutrophils, clearing the marrow - this is the first successful example of differentiation therapy in oncology
- Cure rate with ATRA + ATO: ~85-90%
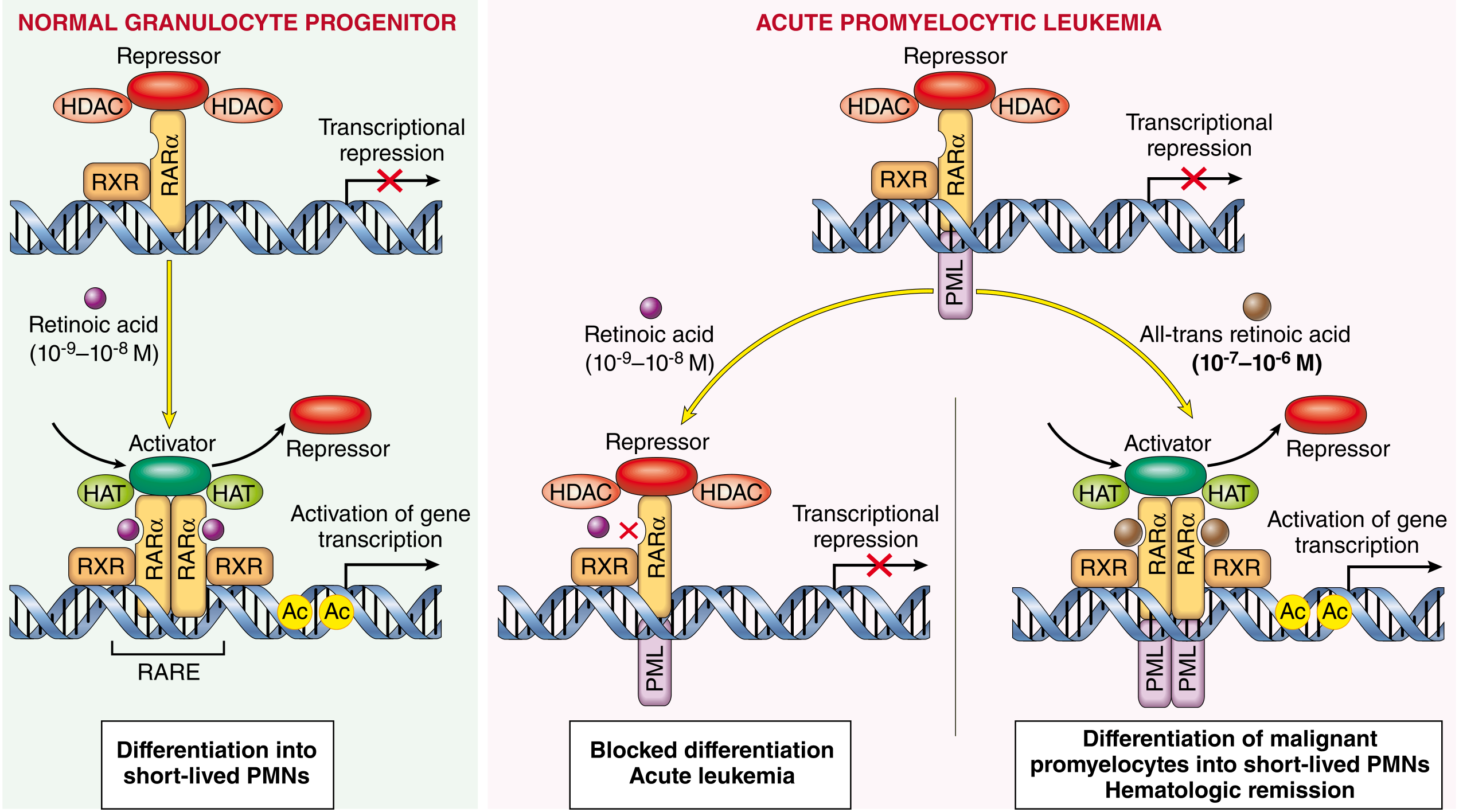
Molecular pathogenesis of APL and basis for response to ATRA. The PML-RARα fusion protein (left) blocks differentiation. At pharmacologic ATRA doses (right), the block is relieved and malignant promyelocytes differentiate into mature, short-lived neutrophils. - Robbins, Cotran & Kumar Pathologic Basis of Disease
4. Measurable Residual Disease (MRD) Monitoring
- Quantitative RT-PCR for fusion transcripts (e.g., PML-RARA, RUNX1-RUNX1T1, CBFB-MYH11) or NPM1 mutations
- Flow cytometry for aberrant immunophenotypes
- MRD positivity after treatment predicts relapse and guides decisions on transplantation
Complications
| Complication | Mechanism | Notes |
|---|---|---|
| Infection / sepsis | Neutropenia | Most common cause of death |
| DIC | Tissue factor release from blasts | Especially APL and monocytic AML |
| Tumor lysis syndrome | Rapid cell breakdown | Hyperuricemia, hyperphosphatemia, hypocalcemia, AKI |
| Hyperleukocytosis / leukostasis | WBC >100,000; blasts aggregate in microvasculature | Respiratory failure, CNS injury |
| Differentiation syndrome (ATRA syndrome) | ATRA/ATO therapy in APL | Fever, pulmonary infiltrates, effusions, weight gain; treated with dexamethasone |
| Cardiotoxicity | Anthracyclines | Dose-dependent, cumulative |
Summary of Key Points
- AML = clonal proliferation of immature myeloid blasts; diagnosis requires ≥20% blasts (exceptions: CBF-AML, APL)
- Pathogenesis involves transcription factor mutations, signaling mutations (FLT3, KIT), and epigenetic dysregulation (IDH1/2, DNMT3A, TET2)
- Classification is now predominantly genetic (WHO 5th Ed. 2022 / ICC 2022)
- APL is uniquely treated with ATRA + ATO (differentiation therapy) with excellent outcomes
- Standard induction: 7+3 (cytarabine + anthracycline); targeted agents added based on molecular profile
- Prognosis is determined primarily by cytogenetics and molecular mutations (ELN risk stratification)
- Allogeneic HCT remains the only potentially curative approach for adverse-risk disease
- Novel targeted agents (FLT3 inhibitors, IDH inhibitors, venetoclax, menin inhibitors) have substantially expanded treatment options
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine 22E (2025); Henry's Clinical Diagnosis and Management by Laboratory Methods; Tietz Textbook of Laboratory Medicine 7th Ed.; Goldman-Cecil Medicine.
This is a shared conversation. Sign in to Orris to start your own chat.