Classify and describe the etiology, pathogenesis, manifestations, radiologic and morphologic features and complications of Paget's disease of the bone
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Loading Image
I now have everything needed for a comprehensive, well-illustrated answer. Let me compile it.
Paget's Disease of Bone (Osteitis Deformans)
Classification
Paget's disease is classified as a focal, metabolic bone disorder characterized by markedly increased but structurally disorganized bone remodeling. It is subdivided by extent:
- Monostotic (~15% of cases): one bone affected
- Polyostotic (~85% of cases): multiple, non-contiguous skeletal sites involved
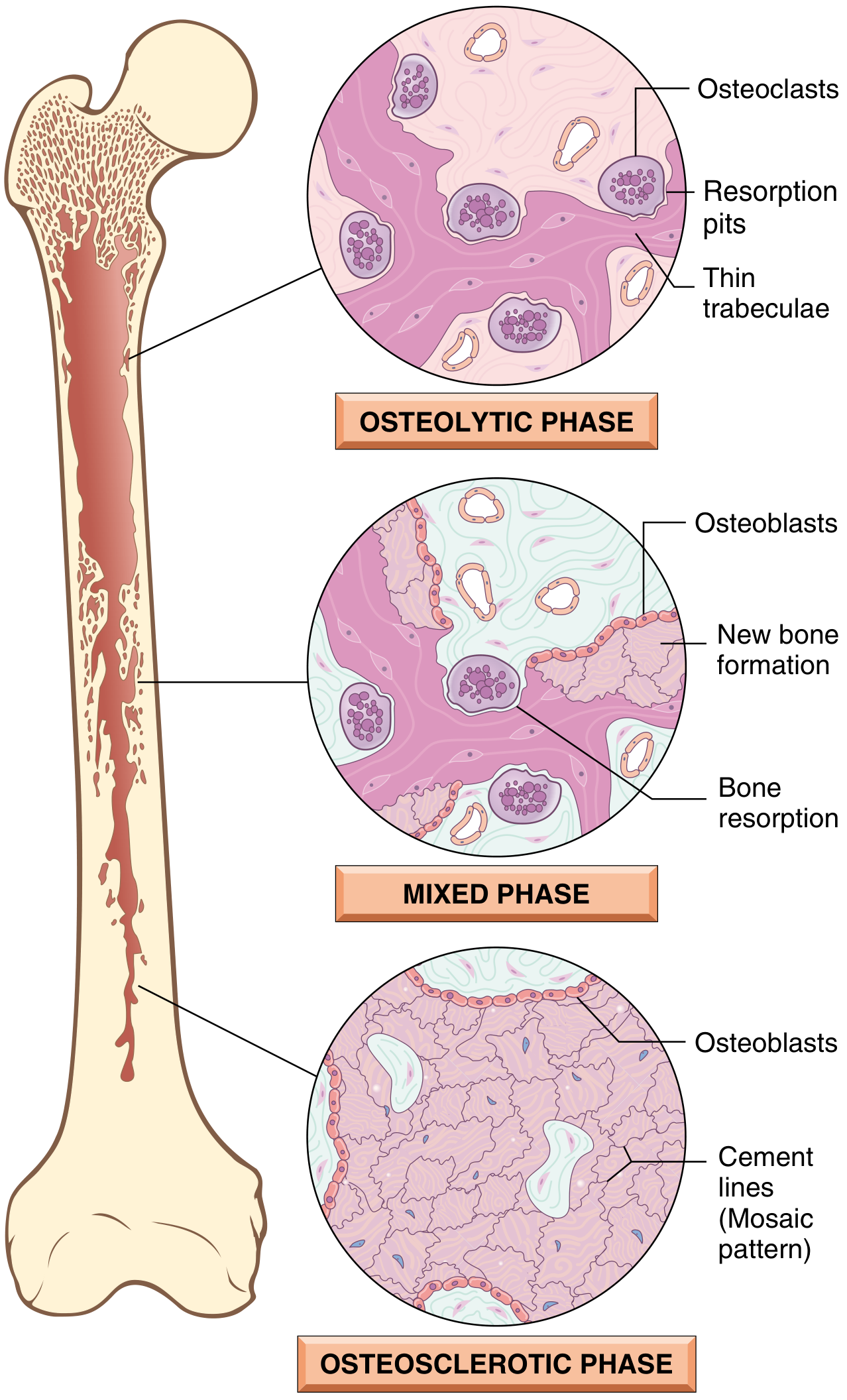
The disease follows three sequential phases:
- Initial osteolytic (resorptive) phase
- Mixed osteoclast-osteoblast phase
- Final osteosclerotic (burned-out) phase
Epidemiology
- Prevalence ~0.5-1% in the UK and US; autopsy studies reveal ~3% in those over age 40
- More common in males (ratio ~1.4-1.7:1), rising with age
- High prevalence in Western Europe (UK, France, Germany), Australia, New Zealand, and among European immigrants
- Rare in Scandinavia, East Asia, sub-Saharan Africa, and native populations of the Americas
- Prevalence and severity have decreased in recent decades for unclear reasons
Etiology
The cause remains incompletely understood; both genetic and environmental factors contribute.
Genetic Factors
- SQSTM1 (sequestosome-1/p62) mutation is the most important: found in ~60% of familial cases and ~10-16% of sporadic cases; more than 20 mutations described in or near the ubiquitin-associated (UBA) domain. p62 is a scaffold protein regulating NF-κB signaling, the master pathway for osteoclast differentiation. Mutations increase NF-κB activity and thus osteoclast activity.
- RANK (TNFRSF11A) activating mutations: cause familial expansile osteolysis and early-onset Paget disease
- OPG (TNFRSF11B) inactivating mutations: loss of osteoprotegerin leads to uncontrolled osteoclastogenesis; homozygous deletions cause juvenile Paget disease (familial idiopathic hyperphosphatasia)
- VCP mutations: cause multisystem proteinopathy - inclusion body myopathy + Paget disease + frontotemporal dementia (± ALS)
- ZNF687 mutations: rare syndrome of Paget disease with giant cell tumors
- PFN1 (profilin-1) mutations: early-onset disease with predisposition to osteosarcoma
- Genome-wide association studies identified 7 additional loci near genes including CSF1 (M-CSF), TM7SF4 (DC-STAMP), OPTN (optineurin), and RIN3 - all converging on osteoclast function and autophagy
- Autosomal dominant transmission with variable penetrance; positive family history in 15-25% of patients (raises prevalence 7-10-fold in first-degree relatives)
Environmental / Viral Factors
- In vitro studies raised the possibility of chronic paramyxoviral infection (measles virus, canine distemper virus, respiratory syncytial virus) of osteoclast precursors - nuclear inclusions in pagetic osteoclasts resemble nucleocapsids of paramyxoviruses
- Geographic variation and declining incidence over time support an environmental trigger
- Mechanical loading, skeletal injuries, rural exposures, and toxins have also been proposed
Pathogenesis
The central defect is abnormal osteoclast behavior driven by the interaction of genetic susceptibility (especially SQSTM1/NF-κB dysregulation) with environmental triggers.
Key mechanisms:
- Osteoclast hyperactivation: pagetic osteoclasts are increased in number, markedly enlarged, and hypernucleated (up to 100 nuclei vs. normal 3-5). They show hypersensitivity to RANKL and 1,25(OH)₂D₃
- RANKL/OPG imbalance: marrow stromal cells from pagetic lesions overexpress RANKL; osteoclast precursor recruitment is amplified by IL-6 (elevated in blood of active disease)
- Osteoblastic response: compensatory osteoblastic new bone formation is markedly increased, exceeding the amount resorbed - producing enlarged, deformed bones
- Woven bone: the new bone is deposited in a disorganized (woven) pattern with impaired mechanical strength
- Autophagy defects: nuclear inclusions in pagetic osteoclasts may represent aggregates of undegraded proteins due to defects in autophagy; proteins encoded by VCP, OPTN, and SQSTM1 all regulate autophagy
- Marrow fibrosis and hypervascularity: increased vascularity causes warmth overlying lesions; marrow is replaced by loose fibrovascular tissue in active phases
- Focal nature: possibly explained by somatic mutations in affected bones causing local osteoclast hyperactivity, or by early-life mechanical injury creating a focus for remodeling
Clinical Manifestations
An estimated 7-16% of patients come to medical attention; ~20% of those are completely asymptomatic, with Paget disease detected incidentally on lab work (elevated ALP) or imaging.
Symptoms
| System | Manifestations |
|---|---|
| Bone pain | Most common symptom; from microfractures, increased bone turnover, or secondary joint disease |
| Skull | Headache, skull enlargement (hat size increase), leontiasis ossea (lion-face), platybasia (invagination of skull base), compression of posterior fossa |
| Cranial nerves | Sensorineural or conductive deafness (most common CN complication; from temporal bone involvement or cochlear nerve compression), vertigo, facial nerve palsy, vision changes |
| Spine | Back pain, spinal stenosis, nerve root compression, kyphosis, radiculopathy, myelopathy |
| Pelvis | Pelvic bowing, hip pain from secondary osteoarthritis, protrusio acetabuli |
| Long bones | Anterior bowing of femur and tibia, leg-length discrepancy, secondary severe osteoarthritis of adjacent joints |
| Skin | Warmth overlying pagetic bones due to hypervascularity |
| Cardiac | High-output heart failure in severe polyostotic disease (increased blood flow acting as AV shunt) |
| Labs | Markedly elevated serum ALP (bone-specific); bone resorption markers (N-telopeptide, C-telopeptide) elevated; serum Ca²⁺ and PO₄³⁻ usually normal |
Skeletal Distribution
- Axial skeleton and proximal femur involved in up to 80% of cases
- Most commonly: pelvis > lumbar spine > femur > thoracic spine > skull > tibia
Radiologic Features
Plain radiographs are usually diagnostic. Key findings include:
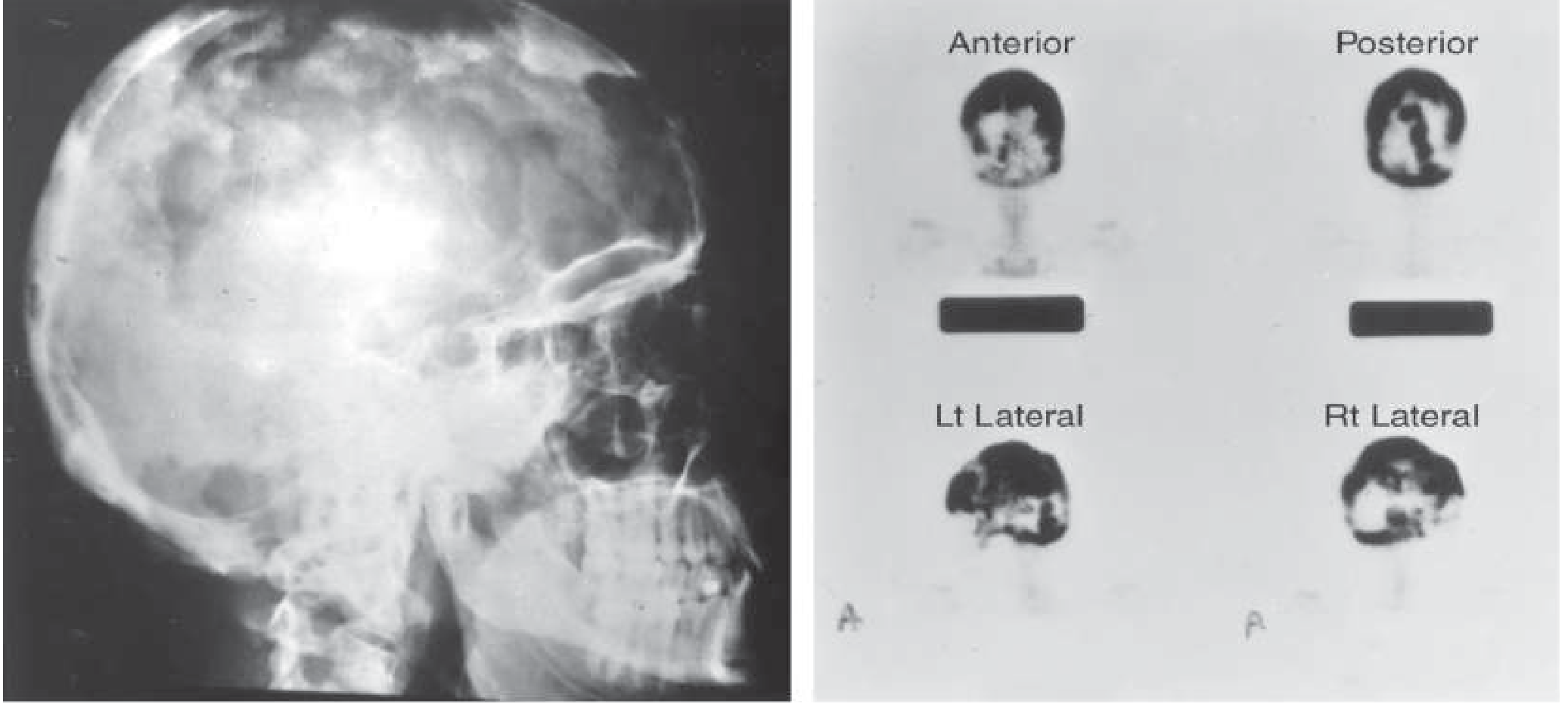
Skull
- "Cotton-wool" appearance: mixed lytic and sclerotic foci
- Osteoporosis circumscripta: well-defined lytic area (early/active phase) in frontal or occipital bone
- Thickening of diploic tables; enlargement and sclerosis of calvarium
- Bone scan: diffuse intense isotope uptake by frontal, parietal, occipital, and petrous bones
Spine
- "Picture-frame" vertebra: cortical thickening of superior and inferior endplates with central lucency
- "Ivory vertebra": diffuse radiodense enlargement of the entire vertebral body
Pelvis
- Disruption or fusion of sacroiliac joints
- Mixed porotic and radiodense lesions of ilium with coarse trabeculation (whorls)
- Brim sign: thickened and sclerotic iliopectineal line
- Softening with protrusio acetabuli; axial migration of hips
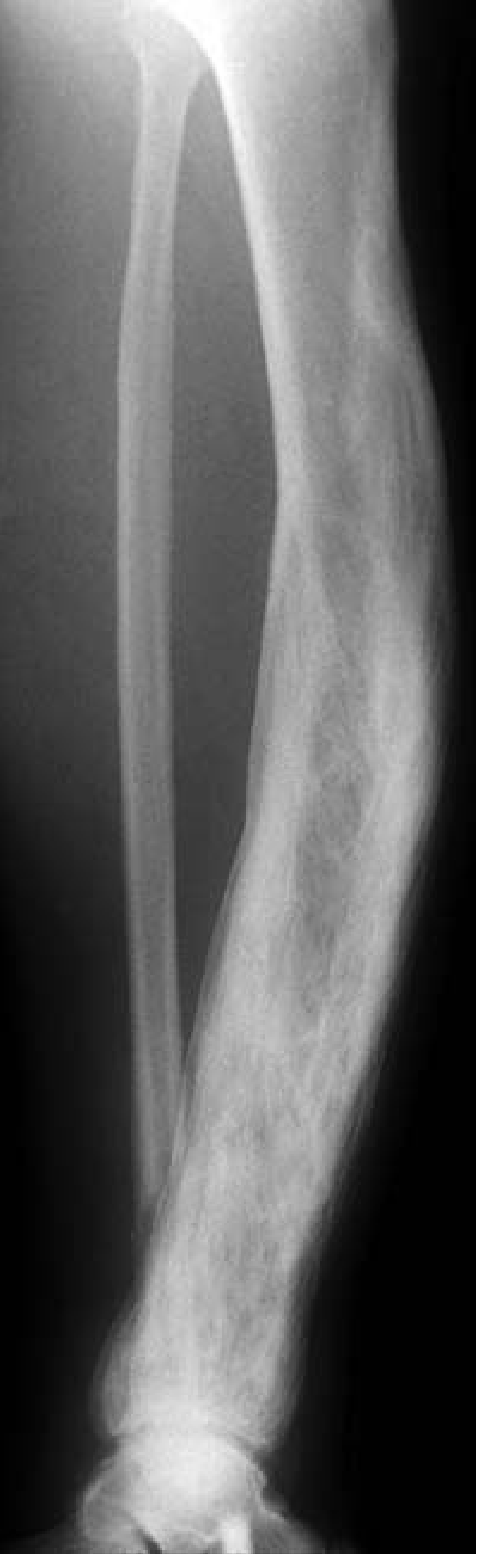
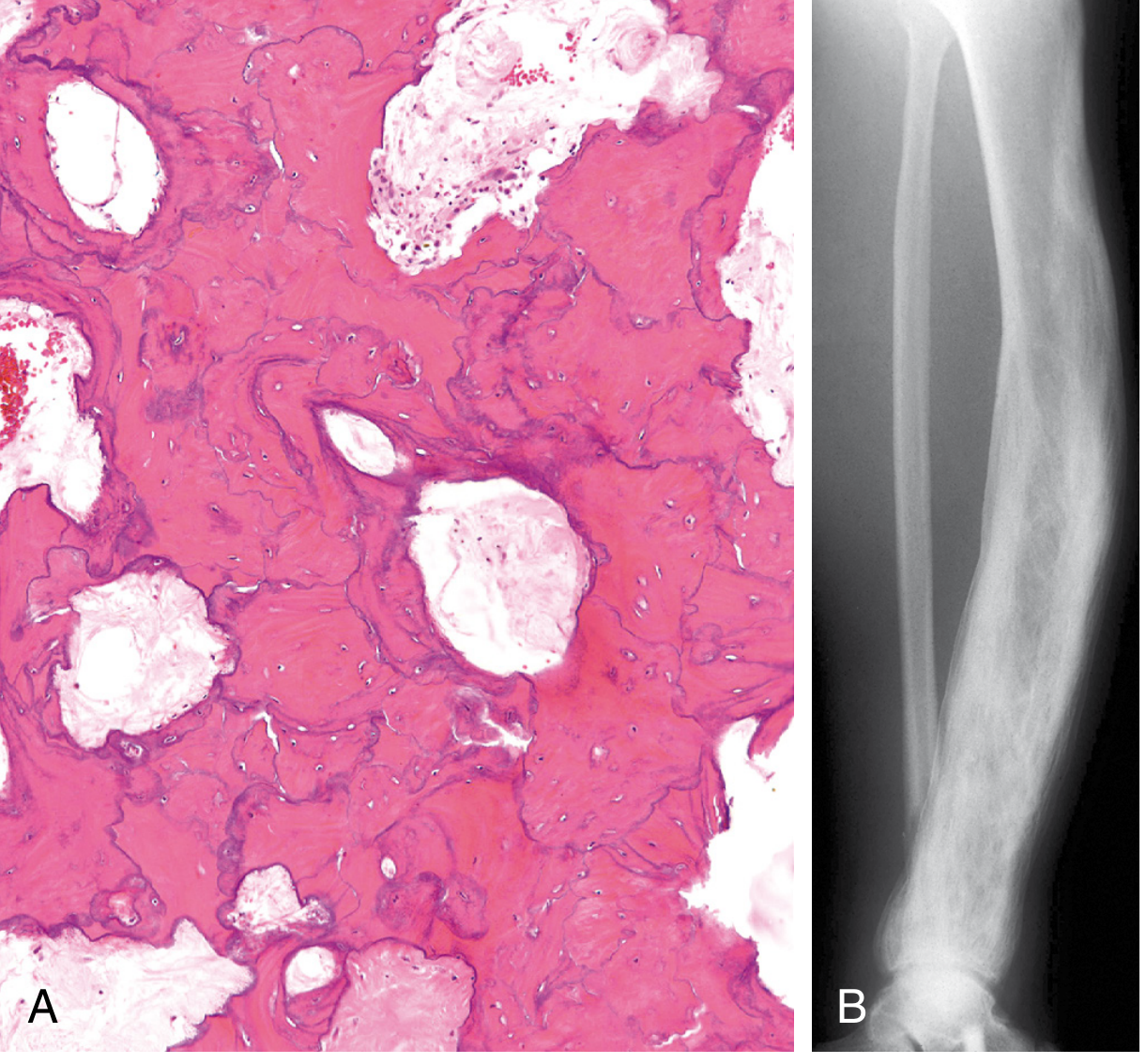
Long Bones
- Enlargement/expansion of the whole bone or involved segment
- Cortical thickening (both periosteal and endosteal)
- Coarsening of trabecular markings
- Bowing deformity (typically anterior in tibia/femur)
- Mixed lucency and sclerosis
- "Blade of grass" or "flame" sign: advancing lytic front at the edge of lesion in active phase
Other Imaging
- ⁹⁹ᵐTc bone scan: most sensitive modality for identifying active lesions and disease extent; less specific than plain films
- CT: useful for assessment of fracture, cortical detail, and nerve compression
- MRI: necessary when sarcomatous transformation, giant cell tumor, or metastatic disease is suspected in pagetic bone
Morphologic Features
Gross Pathology
- Affected bones are enlarged, thickened, and deformed
- Cortices are soft and porous - lack structural stability
- Coarsely thickened trabeculae
- Cut surface shows increased vascularity
Histology - Phase-Dependent
Phase 1: Osteolytic Phase
- Numerous abnormally large osteoclasts with up to 100 nuclei (normally ≤4)
- Prominent resorption pits (Howship's lacunae)
- Thin trabeculae being actively destroyed
- Some osteoclasts contain intranuclear inclusions (paramyxovirus-like)
Phase 2: Mixed Phase
- Both osteoclasts and osteoblasts active simultaneously
- Osteoblastic new bone formation alongside continued resorption
- Poorly mineralized woven bone being laid down
- Marrow replaced by loose fibrovascular connective tissue rich in osteoprogenitor cells and blood vessels
- Large osteoclasts visible close to areas of new bone formation; extensive marrow fibrosis
Phase 3: Osteosclerotic (Burned-out) Phase
- Hallmark: mosaic pattern of lamellar bone (jigsaw puzzle-like)
- Produced by unusually prominent, haphazardly oriented cement lines joining units of lamellar bone in all directions
- Cell activity decreases; fibrovascular tissue recedes and is replaced by normal marrow
- Coarsely thickened trabeculae; soft, porous cortices
Complications
1. Fractures
- "Chalk-stick" (transverse) fractures: characteristic of Paget disease; typically involve weight-bearing long bones (femur, tibia)
- Result from structurally inferior woven bone and cortical softening
- Vertebral compression fractures can cause kyphosis or spinal cord injury
2. Secondary Osteoarthritis
- Most common joint affected: hip (from femoral head deformity and protrusio acetabuli)
- Also knee and spine; results from bony deformity distorting joint mechanics
3. Neurologic Complications
- Sensorineural and conductive deafness: most common; temporal bone involvement affecting the ossicles and cochlea
- Spinal stenosis and radiculopathy from vertebral enlargement
- Cranial nerve palsies (VII, II)
- Platybasia with posterior fossa compression
- Spinal cord compression and myelopathy
4. Neoplastic Transformation (most feared)
- Secondary osteosarcoma: occurs in <1% of all Paget disease patients overall, but 5-10% of those with severe polyostotic disease; rapidly fatal with very poor prognosis
- Other tumors: giant cell tumor (especially in ZNF687 mutation carriers), fibrosarcoma, chondrosarcoma
- Suspect when a previously stable lesion rapidly enlarges or becomes increasingly painful
5. Cardiovascular Complications
- High-output cardiac failure: rare; seen in severe polyostotic disease where increased bone vascularity functions as an arteriovenous shunt
- May exacerbate underlying coronary artery disease
6. Hypercalcemia
- Typically only during immobilization (when osteoclastic resorption is uncoupled from osteoblastic deposition) - may precipitate nephrolithiasis
7. Systemic Proteinopathy (VCP mutations)
- Inclusion body myopathy + frontotemporal dementia + ALS in addition to bone disease
Biochemical Markers
| Marker | Significance |
|---|---|
| Serum ALP (total) | Test of choice - markedly elevated; reflects extent and severity; used for monitoring treatment response |
| Bone-specific ALP / PINP | Useful when total ALP normal but disease active at one site |
| Serum/urine N-telopeptide, C-telopeptide | Bone resorption markers; elevated in active disease; fall faster than ALP with treatment |
| Serum Ca²⁺, PO₄³⁻ | Usually normal (unless immobilized) |
| Osteocalcin | Not reliably elevated; not recommended |
Summary
Paget's disease of bone is a focal remodeling disorder driven primarily by pathologic osteoclast overactivation, most commonly linked to SQSTM1/NF-κB pathway dysregulation and possible paramyxoviral environmental triggers. It evolves through three histologic phases (lytic → mixed → sclerotic), producing the pathognomonic mosaic cement-line pattern on histology. Radiologically, it shows characteristic bone expansion, cortical thickening, mixed lysis/sclerosis, bowing deformities, and site-specific patterns (cotton-wool skull, picture-frame vertebra, brim sign in pelvis). Complications range from fractures and osteoarthritis to nerve compression and, most seriously, osteosarcomatous transformation in severe polyostotic disease.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528, p. 1090-1091) | Harrison's Principles of Internal Medicine 22E (9781265977061, p. 3355-3358) | Goldman-Cecil Medicine (9780323930345, p. 2651-2653) | Firestein & Kelley's Textbook of Rheumatology (9780323935364) | Rheumatology 2-Volume Set, Elsevier (9780702081330, p. 572-691)
This is a shared conversation. Sign in to Orris to start your own chat.