Barter syndrome etiology
Reading File
Finding Sources
Finding Sources
Searching PubMed
Bartter syndrome genetics etiology
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Now I have everything needed for a thorough, well-illustrated answer.
Bartter Syndrome - Etiology
Bartter syndrome is a group of autosomal recessive hereditary tubulopathies caused by loss-of-function mutations in genes encoding transport proteins in the thick ascending limb (TAL) of the loop of Henle. All subtypes share the same final common pathway: impaired NaCl reabsorption in the TAL, leading to salt wasting, volume contraction, secondary hyperreninemic hyperaldosteronism, hypokalemic hypochloremic metabolic alkalosis, and elevated prostaglandin production.
Pathophysiological Basis
Normal NaCl reabsorption in the TAL depends on a coordinated set of transporters (shown below):
- Apical (lumen-facing): NKCC2 cotransporter brings Na+/K+/2Cl- into the cell; ROMK recycles K+ back into the lumen to sustain NKCC2 activity and generate a lumen-positive voltage that drives paracellular reabsorption of Na+, Ca2+, and Mg2+
- Basolateral: CLC-Kb (+ its subunit Barttin) extrudes Cl- out; CaSR on the basolateral surface tonically inhibits ROMK
A mutation in any of these components abolishes TAL NaCl absorption.
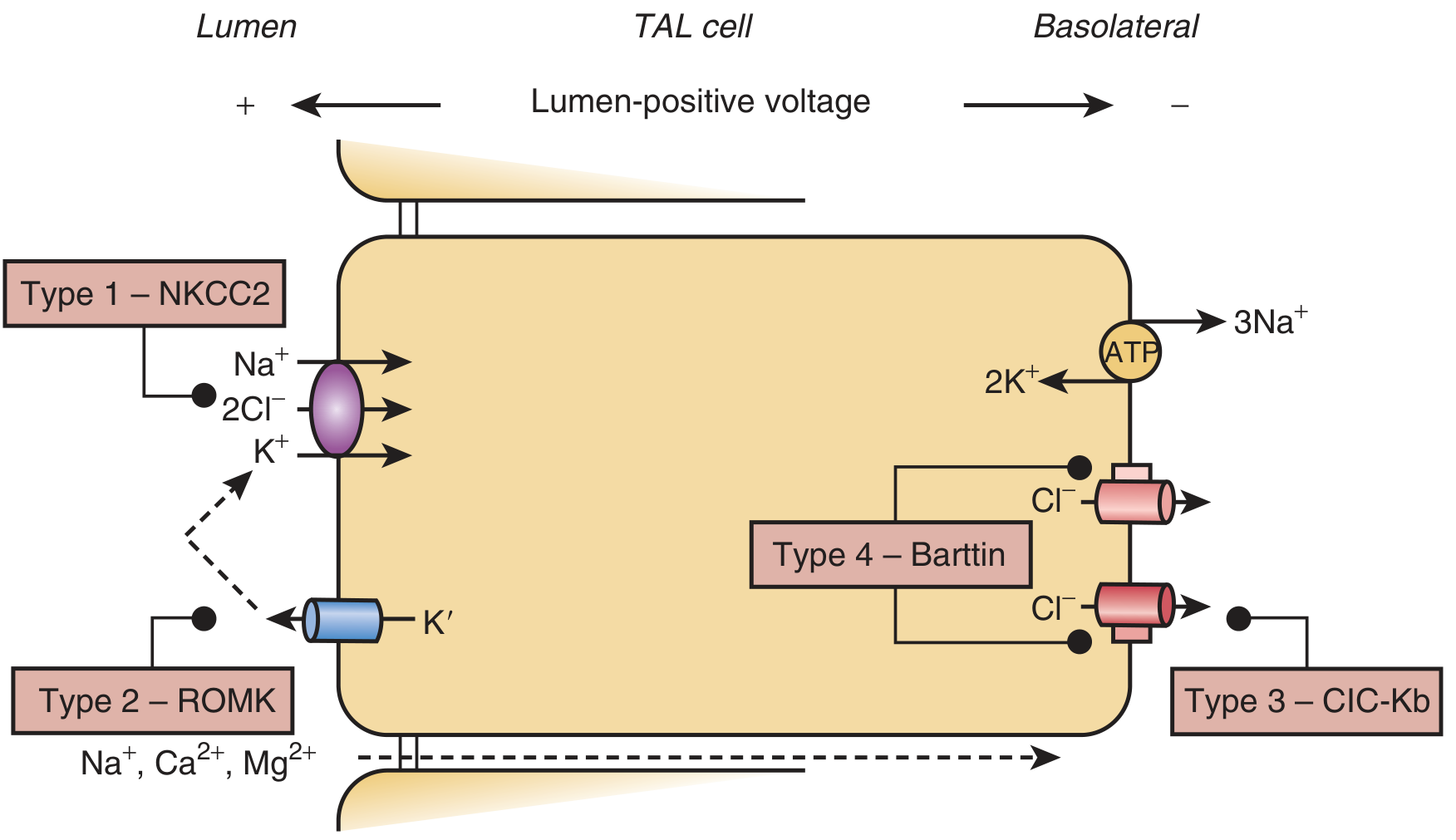
Fig. Schematic of the TAL cell showing which transporter is disrupted in each Bartter type. (Brenner & Rector's The Kidney)
Genetic Types
| Type | Gene | Protein | Clinical Presentation |
|---|---|---|---|
| Type I | SLC12A1 | NKCC2 (apical Na-K-2Cl cotransporter) | Antenatal - polyhydramnios, premature birth, severe salt wasting, hypercalciuria, nephrocalcinosis |
| Type II | KCNJ1 | ROMK (apical K+ channel) | Antenatal - same as type I; paradoxical transient neonatal hyperkalemia (ROMK is also needed for K+ secretion in collecting duct) |
| Type III | CLCNKB | CLC-Kb (basolateral Cl- channel) | Classical Bartter - childhood/adolescent onset, milder, rarely nephrocalcinosis; type CLCNKA can partially compensate |
| Type IV | BSND | Barttin (accessory subunit of both CLC-Ka and CLC-Kb) | Antenatal + sensorineural deafness; Barttin is expressed in inner ear epithelia; loss disables both chloride channels |
| Type IVb | CLCNKA + CLCNKB | Both CLC-Ka and CLC-Kb | Severe antenatal phenotype with deafness - equivalent to type IV |
| Type V | CASR (activating mutation) | Calcium-sensing receptor (CaSR) | Gain-of-function mutation (unlike all others); activated CaSR inhibits ROMK on basolateral surface, suppressing TAL transport; associated with autosomal dominant hypocalcemia (ADH) |
Key Details by Type
Types I & II - Antenatal Bartter
- The most severe forms, presenting in utero or immediately after birth
- Polyhydramnios (fetal polyuria), premature delivery, postnatal polyuria, vomiting, failure to thrive
- Prominent hypercalciuria and nephrocalcinosis
- Impaired chloride sensing at the macula densa disrupts tubuloglomerular feedback (TGF), leading to primary activation of COX-2 with excess prostaglandin E2 production - this is why COX inhibitors (indomethacin) are therapeutic
- Type II has a unique neonatal transient hyperkalemia because ROMK is also expressed in the collecting duct and is needed for K+ secretion there
Type III - Classical Bartter
- Presents in childhood or adolescence with failure to thrive, polyuria, polydipsia
- Hypokalemia, hypochloremic metabolic alkalosis, hyperreninemia, hyperaldosteronism
- Hypercalciuria in ~80%; ~20% have hypomagnesemia
- Milder because CLC-Ka (encoded by CLCNKA) can partially compensate for lost CLC-Kb function - but when both CLCNKA and CLCNKB are mutated (Type IVb), the phenotype is severe/antenatal
- Up to one-third of type 3 patients can actually present antenatally or in the neonatal period despite the "classical" designation
Type IV - Bartter with Sensorineural Deafness
- Barttin is co-expressed with CLC-Ka and CLC-Kb in the stria vascularis of the cochlea, where it is required for K+ recycling in endolymph generation
- Loss of Barttin function abolishes both chloride channels in the TAL and the inner ear simultaneously, producing the combination of severe Bartter syndrome and bilateral sensorineural deafness
Type V - CaSR Activating Mutation
- Unlike all other types, this is a gain-of-function mutation (dominant), not a loss-of-function
- Activated CaSR tonically inhibits ROMK, thus reducing TAL NaCl reabsorption
- Presents with metabolic alkalosis, hypokalemia, hypercalciuria, hypocalcemia, and low PTH - the "Bartter syndrome phenotype" in the context of autosomal dominant hypocalcemia
Downstream Consequences (Common to All Types)
- Failure of TAL NaCl reabsorption → urinary salt wasting → extracellular volume contraction
- Volume contraction → activation of renin-angiotensin-aldosterone system (secondary hyperreninemic hyperaldosteronism)
- Increased NaCl delivery to the collecting duct → aldosterone-driven K+ and H+ secretion → hypokalemia and metabolic alkalosis
- Impaired TGF at macula densa → COX-2 upregulation → excess prostaglandin E2 → further afferent arteriolar dilation, augmented renin release, and juxtaglomerular hypertrophy
- Vascular unresponsiveness to angiotensin II (despite markedly elevated renin/aldosterone)
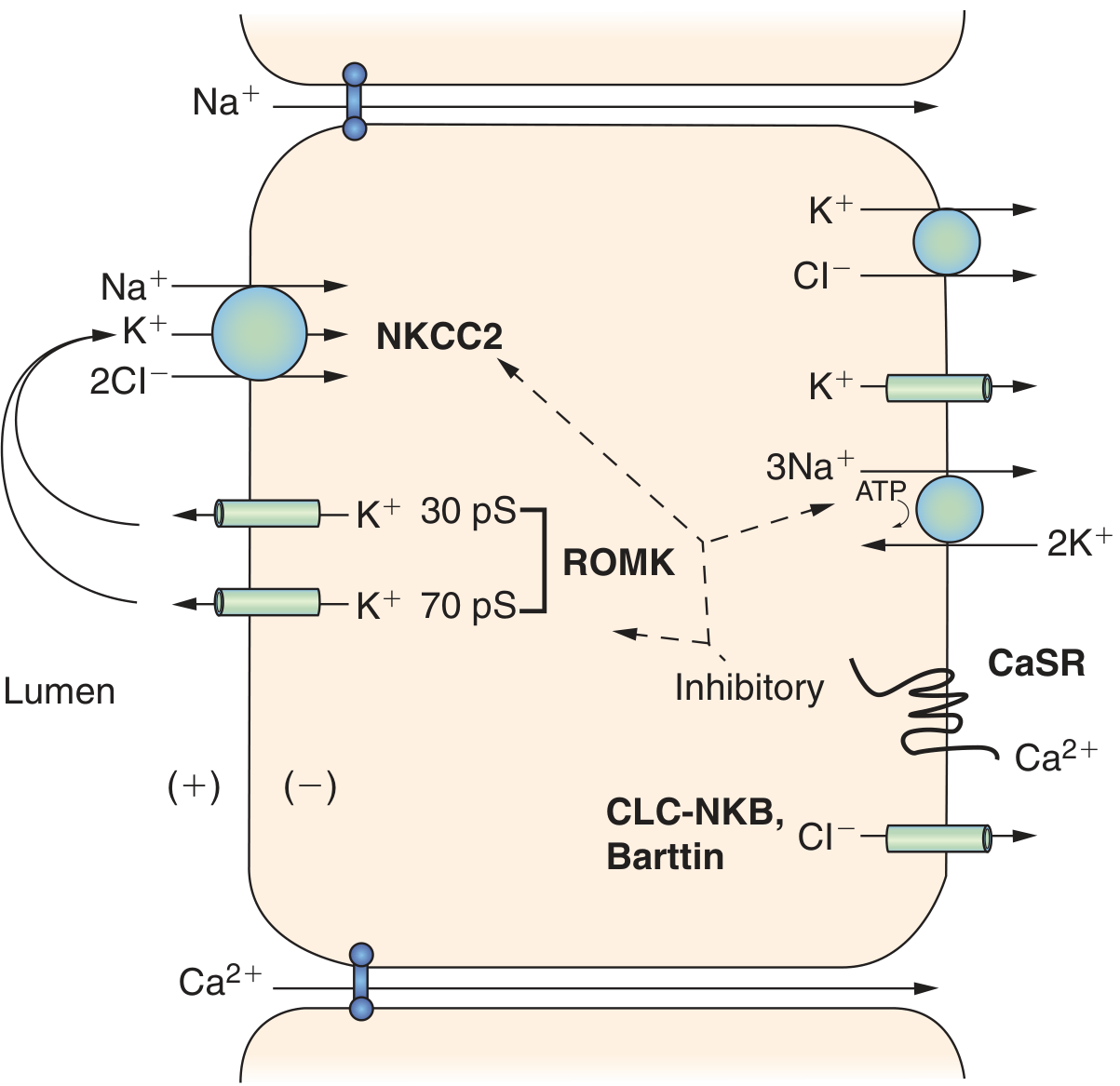
Fig. The CaSR inhibitory effect on ROMK in the TAL; relevant to Type V Bartter. (Brenner & Rector's The Kidney)
Inheritance & Genetics Summary
- Types I-IV: Autosomal recessive - patients are typically homozygotes or compound heterozygotes
- Type V: Autosomal dominant (activating CaSR mutation)
- A minority of patients with clear clinical Bartter syndrome have no identifiable mutation in any of the five known genes - unexplained mutations in other genes or dominant-negative effects are hypothesized
Sources:
- Brenner and Rector's The Kidney, 2-Volume Set, Chapters 72 & 73
- National Kidney Foundation Primer on Kidney Diseases, 8th Edition, Chapter 14
This is a shared conversation. Sign in to Orris to start your own chat.