Pediatric leukodystrophy
Reading File
Finding Sources
Searching the Web
pediatric leukodystrophy classification diagnosis treatment 2024 2025
Searching PubMed
leukodystrophy pediatric
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
I now have rich content from multiple authoritative textbooks. Let me compile the comprehensive answer.
Pediatric Leukodystrophy
Leukodystrophies are a group of >50 inherited neurological disorders that impair the development or maintenance of central nervous system (CNS) myelin. As a group, their combined incidence is at least 1 in 4,700 live births (some estimates: 1 in 7,663 per Bonkowski et al., 2010), making them a significant cause of pediatric neurological morbidity and death.
Definition and Core Concept
The term "leukodystrophy" (Greek: leuko = white, dystrophy = abnormal growth/maintenance) refers to disorders where the primary defect lies in the white matter - specifically in myelin production or maintenance. The pathology can affect:
- Oligodendrocytes (CNS myelin-forming cells)
- Schwann cells (PNS myelin)
- Astrocytes (as in Alexander disease)
- Peroxisomal or lysosomal enzymes critical for myelin lipid metabolism
Classification
Leukodystrophies are broadly grouped by their underlying mechanism:
| Category | Examples |
|---|---|
| Lysosomal storage disorders | MLD (arylsulfatase A deficiency), Krabbe disease (galactocerebrosidase deficiency) |
| Peroxisomal disorders | X-linked adrenoleukodystrophy (X-ALD), Zellweger spectrum |
| Myelin structural protein defects | Pelizaeus-Merzbacher disease (PLP1 gene) |
| Astrocyte dysfunction | Alexander disease (GFAP mutation) |
| eIF2B / integrated stress response | Vanishing white matter disease |
| Mitochondrial | MERRF, MELAS (overlap) |
| Aminoacidopathy | Canavan disease (aspartoacylase deficiency) |
Key Diseases in Detail
1. Metachromatic Leukodystrophy (MLD)
-
Gene/Enzyme: ARSA gene (chromosome 22q13) → arylsulfatase A (ASA) deficiency; rarely PSAP gene → saposin B deficiency
-
Inheritance: Autosomal recessive
-
Pathophysiology: Sulfatide accumulates in CNS/PNS myelin, causing progressive demyelination; affects both central and peripheral myelin
-
Subtypes by age of onset:
- Late infantile (6 months-2 years) - most common; gait disorder, hypotonia, lower-limb areflexia early; CNS involvement follows
- Juvenile (3-16 years) - intellectual impairment, behavioral difficulties, ataxia, upper motor neuron signs, peripheral neuropathy
- Adult - dementia, psychiatric features (psychosis, hallucinations), dementia; subtle neuropathic signs
-
Diagnosis:
- MRI: large confluent symmetrical T2-hyperintense areas in cerebral white matter, brainstem, cerebellum
- Elevated urinary sulfatide excretion
- Reduced ASA enzyme in leukocytes/fibroblasts (confirmed by molecular testing, since pseudodeficiency exists)
- Elevated CSF protein; reduced nerve conduction velocities
-
Treatment:
- LENMELDY (atidarsagene autotemcel) - FDA-approved in 2024; ex vivo lentiviral HSC gene therapy using the patient's own stem cells transduced with a healthy ARSA gene. Indicated for pre-symptomatic or early-symptomatic late infantile/early juvenile MLD
- Hematopoietic stem cell transplantation (HSCT) can slow progression in pre-symptomatic patients
- Long-term follow-up data (Fumagalli et al., NEJM 2025) support durability of gene therapy
-
Bradley and Daroff's Neurology in Clinical Practice; Goldman-Cecil Medicine
2. Krabbe Disease (Globoid Cell Leukodystrophy)
-
Gene/Enzyme: GALC gene (chromosome 14q31) → galactocerebrosidase (galactocerebroside β-galactosidase) deficiency
-
Inheritance: Autosomal recessive
-
Pathophysiology: Galactocerebroside and psychosine (galactosylsphingosine) accumulate → oligodendrocyte destruction → marked demyelination; characteristic globoid cells (multinucleated macrophages filled with galactocerebroside) in cerebral white matter
-
Clinical features:
- Early infantile (classic): Onset 3-6 months; rapid deterioration, hypertonicity, opisthotonic posturing, optic atrophy, seizures, peripheral neuropathy
- Late-onset: Peripheral neuropathy and spasticity may be the only features
-
Diagnosis: Galactocerebrosidase enzyme assay; NBS programs use lyso-Gb1 as a biomarker; MRI shows symmetric white matter involvement; very slow nerve conduction velocities
-
Treatment: HSCT/cord blood transplantation if performed before symptom onset (newborn screening urgency); no disease-modifying treatment once symptomatic
-
Bradley and Daroff's Neurology in Clinical Practice; Goldman-Cecil Medicine
3. X-Linked Adrenoleukodystrophy (X-ALD)
-
Gene/Enzyme: ABCD1 gene (Xq28) → ALDP (peroxisomal membrane protein) deficiency → VLCFA accumulate (not transported into peroxisomes for beta-oxidation)
-
Inheritance: X-linked recessive
-
Prevalence: ~1 in 20,000 - the most common peroxisomal disorder
-
Phenotypes in males:
- Childhood cerebral ALD (CCALD): Onset 4-8 years; deteriorating school performance, behavioral changes, vision deficits, adrenal insufficiency; symmetrical white matter lesions on MRI; rapidly progressive to total disability and death
- Adrenomyeloneuropathy (AMN): Adult onset; progressive spastic paraparesis, cerebral demyelination, adrenal insufficiency
- Isolated adrenal insufficiency (Addison's disease): ~20% of males
- Carrier females may develop symptoms (spastic paraparesis, sphincter dysfunction) but are typically spared from cerebral ALD and adrenal insufficiency
-
Diagnosis: Elevated plasma very-long-chain fatty acids (VLCFAs), especially C26:0 and C26:0/C22:0 ratio; ABCD1 mutation confirmation; MRI brain
-
Monitoring: Cortisol stimulation testing every 6-9 months for adrenal insufficiency in all males; regular MRI surveillance
-
Treatment:
- Adrenal insufficiency: oral corticosteroid supplementation (life-saving)
- CCALD before MRI lesion progression: HSCT or hematopoietic stem cell gene therapy (Skysona/elivaldogene autotemcel - approved in Europe)
- "Lorenzo's oil" (dietary therapy): has proved disappointing for reversing neurological disease but may slow VLCFA accumulation in asymptomatic boys
-
Tietz Textbook of Laboratory Medicine; Emery's Elements of Medical Genetics and Genomics
4. Pelizaeus-Merzbacher Disease (PMD)
-
Gene: PLP1 gene (X-linked) → proteolipid protein deficiency or dysfunction
-
Inheritance: X-linked recessive
-
Onset: 3 months to 9 years
-
Clinical features: Slowly progressive myelopathy, nystagmus (pendular), cerebellar involvement, spasticity, cognitive impairment; death between 6-25 years in classic form; milder forms (spastic paraplegia type 2) recognized in adults
-
Variants: PMD-like disease type 1 and spastic paraplegia type 44 caused by GJC2 mutations (autosomal recessive)
-
Diagnosis: MRI (hypomyelination pattern - T2 signal does not suppress white matter); genetic testing for PLP1 mutations
-
Treatment: Supportive only; no specific therapy
-
Goldman-Cecil Medicine
5. Canavan Disease
-
Gene/Enzyme: ASPA gene → aspartoacylase deficiency → N-acetylaspartate (NAA) accumulates → brain edema and dysmyelination
-
Inheritance: Autosomal recessive (enriched in Ashkenazi Jewish population)
-
Clinical features: Developmental delay appearing in first months of life, macrocephaly, hypotonia progressing to spasticity, seizures; diffuse symmetrical white matter degeneration involving subcortical areas and globus pallidus on MRI
-
Diagnosis: Elevated NAA on urine organic acids or MR spectroscopy; ASPA gene testing
-
Treatment: No approved treatment; gene therapy trials are ongoing
-
Goldman-Cecil Medicine
6. Alexander Disease
-
Gene: GFAP (glial fibrillary acidic protein) - gain-of-function mutation
-
Inheritance: Usually de novo (sporadic dominant)
-
Pathophysiology: Mutant GFAP accumulates → abundant Rosenthal fibers in periventricular, perivascular, and subpial locations (hallmark)
-
Subtypes:
- Type I (early/infantile): Seizures, motor deficits, macrocephaly, hydrocephalus from CSF pathway obstruction (requires neurosurgical monitoring); frontal-predominant white matter changes on MRI
- Type II (late onset, older child/adult): Bulbar/pseudobulbar features, ataxia, sleep apnea, autonomic dysfunction; spinal cord involvement
-
Diagnosis: MRI (frontal T2 signal with periventricular rim); GFAP mutation; elevated GFAP in CSF/serum (biomarker)
-
Treatment: Supportive; antisense oligonucleotide (ASO) therapy targeting GFAP mRNA is in clinical trials
-
Robbins Pathologic Basis of Disease; PMC5712822
7. Vanishing White Matter Disease (VWM)
-
Gene: EIF2B1-5 genes → eIF2B proteins (involved in integrated cellular stress response)
-
Inheritance: Autosomal recessive
-
Clinical features: Broad spectrum; from rapidly progressive infantile forms to slowly progressive adult disease; characteristic stress-triggered episodes (episodes triggered by fever or minor trauma causing acute/subacute neurological deterioration); vacuolated myelin, cystic appearance on MRI
-
Treatment: Avoidance of triggers (fever, stress, head trauma); no specific therapy; research into the integrated stress response pathway is active
-
Goldman-Cecil Medicine
Clinical Presentation: Red Flags for Leukodystrophy
| Age of Presentation | Common Features |
|---|---|
| Neonatal/early infancy | Hypotonia, seizures, dysmorphic features (Zellweger), failure to achieve milestones |
| Late infancy (6-24 months) | Developmental regression after a period of normal development, gait disturbance, hypotonia |
| Childhood (2-10 years) | Cognitive/behavioral decline, school failure, spasticity, ataxia, visual loss |
| Adolescence/adulthood | Progressive spastic paraparesis, psychiatric features, cognitive decline |
Key features that should specifically raise suspicion:
- Developmental regression (not just delay)
- Progressive gait disorder + areflexia in young children (MLD, Krabbe)
- Adrenal insufficiency in boys + neurological decline (X-ALD)
- Macrocephaly + megalencephalic leukoencephalopathy patterns
- White matter changes on MRI that are confluent, symmetric, and periventricular
Diagnosis: Stepwise Approach
- Clinical history and exam: Age of onset, regression vs. delay, family history (consanguinity, ethnicity), adrenal symptoms, peripheral neuropathy
- MRI brain (with spectroscopy): Pattern of white matter involvement guides specific diagnosis; some leukodystrophies have near-pathognomonic MRI patterns
- Metabolic screen:
- Plasma VLCFAs (X-ALD)
- Enzyme assays in leukocytes/fibroblasts (arylsulfatase A, galactocerebrosidase, etc.)
- Urine organic acids (elevated NAA in Canavan; sulfatides in MLD)
- Genetic/genomic testing: Gene panel or whole exome sequencing (WES) - now preferred as first-line for undiagnosed cases
- Newborn screening (NBS): Several states/countries now screen for X-ALD (lysophosphatidylcholine C26:0) and Krabbe (galactocerebrosidase activity); MLD NBS programs are emerging
Treatment Overview
| Disease | Available Treatment |
|---|---|
| MLD | LENMELDY (gene therapy, FDA 2024); HSCT (pre-symptomatic) |
| X-ALD | HSCT or gene therapy (Skysona) for early CCALD; steroids for adrenal insufficiency |
| Krabbe | HSCT (pre-symptomatic only - NBS is critical) |
| PMD | Supportive only |
| Canavan | Gene therapy trials (no approved therapy) |
| Alexander | Supportive; ASO trials |
| VWM | Stress avoidance; supportive |
General supportive measures (all leukodystrophies):
- Antiepileptic medications for seizures
- Spasticity management (baclofen, botulinum toxin, orthopedic care)
- Nutritional support (gastrostomy if severe dysphagia)
- Physical, occupational, speech therapy
- Palliative/hospice care planning
- Psychological and family support
Recent Evidence (2024-2026)
- LENMELDY (atidarsagene autotemcel) received FDA approval in 2024 for early-onset MLD; long-term NEJM 2025 data confirm durable benefit (Fumagalli et al., NEJM 2025 [PMID 40577679])
- 2024 European consensus recommendations on MLD NBS and clinical management were published
- A 2025 systematic review on POLR3-related leukodystrophies (Michell-Robinson et al., HGG Adv 2025 [PMID 40684265]) provides comprehensive genotype-phenotype data for this subgroup
- Gene therapy platforms (lentiviral HSC, AAV intrathecal, ASOs) represent the most active therapeutic frontier
Key Practical Takeaways (AAP Clinical Report)
- Leukodystrophies are collectively common enough that general pediatricians must be familiar with them
- Some leukodystrophies require urgent treatment - stage of disease determines efficacy (especially Krabbe and MLD)
- Developmental regression - not just delay - is the cardinal warning sign
- NBS for X-ALD and Krabbe is available and critical for treatment timing
- MRI and metabolic/genetic testing should be pursued promptly when a leukodystrophy is suspected
In the context of a radiology pg
Reading File
Finding Sources
Searching the Web
pediatric leukodystrophy MRI patterns radiology diagnosis white matter 2024
Reading File
Finding Sources
Reading Web Page
https://pmc.ncbi.nlm.nih.gov/articles/PMC10885080
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
Finding Sources
Reading File
Loading Image
Reading File
Reading File
Loading Image
I now have rich radiology-specific content including actual MRI images. Let me now compile the comprehensive radiology PG-focused answer.
Pediatric Leukodystrophy: A Radiology PG Perspective
The radiologist's role in leukodystrophy is central - MRI is the primary modality for detection, characterization, and follow-up. The key skill is pattern recognition: the distribution, signal characteristics, enhancement behavior, and associated findings on MRI allow you to narrow the differential significantly even before biochemistry returns.
The Pattern-Based Approach (PG Must-Know Framework)
Four broad MRI patterns serve as the entry point:
| Pattern | Key Examples |
|---|---|
| Central/periventricular white matter predominant | MLD, Krabbe, X-ALD (posterior), PMD |
| Subcortical U-fiber predominant | Canavan, MLC, L-2-hydroxyglutaricaciduria |
| Frontal predominant | Alexander disease |
| Posterior predominant | X-ALD (childhood cerebral), Krabbe |
Additional clues:
- Enhancement at the leading edge of demyelination → X-ALD (active inflammatory zone)
- Tigroid/leopard skin pattern → MLD
- Macrocephaly on imaging → Alexander, Canavan, MLC
- Basal ganglia involvement → Krabbe (thalami), Canavan (globus pallidus), Alexander (basal ganglia + thalami)
- U-fiber sparing → MLD, Krabbe (early), X-ALD
- Cystic degeneration → VWM, MLC, late Alexander
Disease-by-Disease MRI Features
1. X-Linked Adrenoleukodystrophy (X-ALD)
The most high-yield leukodystrophy for a radiology exam.
- Location: Posterior > anterior; starts at peritrigonal / splenial region (posterior parietal-occipital white matter and splenium of corpus callosum); progresses anteriorly over time
- Signal: Confluent symmetric T2/FLAIR hyperintensity in posterior periventricular white matter; T1 hypointensity in same regions
- Pathognomonic feature: Three-zone pattern (Loes staging):
- Zone 1 (innermost): central necrosis/gliosis - T1 low, T2 high, no enhancement
- Zone 2 (middle): active demyelination - T2 high, enhances (breakdown of blood-brain barrier, active inflammation)
- Zone 3 (outermost/leading edge): early demyelination - T2 slightly elevated
- Enhancement: Rim/marginal enhancement at the leading edge - characteristic and important for staging
- Corpus callosum: Splenium involved early; involvement bridges across midline
- Loes MRI Severity Score (0-34): Used to stage disease and guide treatment decisions (HSCT indicated when score < 9)
- Adrenal: Look for small adrenal glands on abdominal MRI/CT
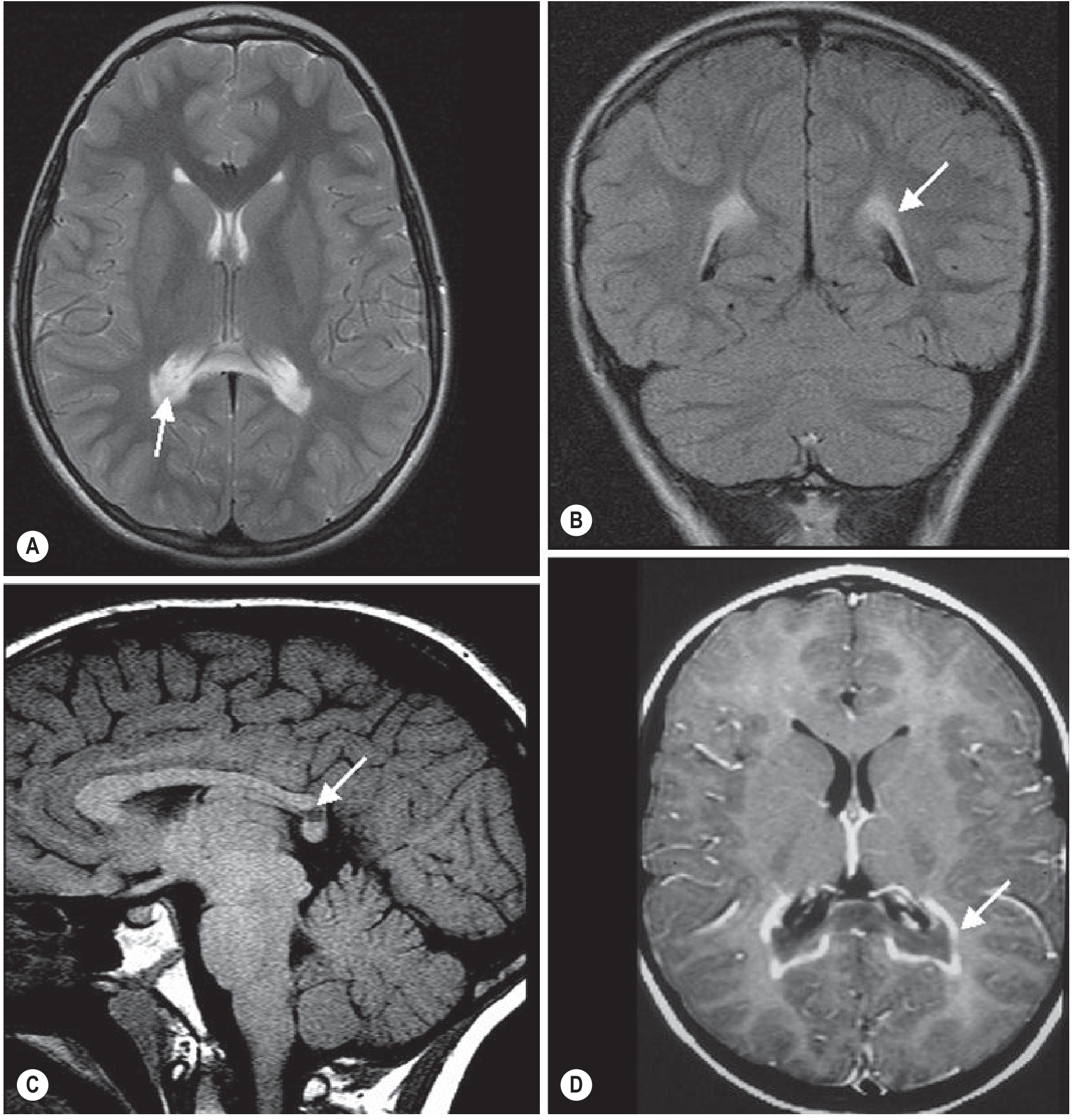
2. Metachromatic Leukodystrophy (MLD)
- Location: Bilateral, symmetric periventricular and central white matter; frontal and parietal predominance; corpus callosum involved (especially splenium); internal capsule and corticospinal tracts involved; cerebellar white matter also affected
- U-fibers: Spared initially (subcortical U-fibers preserved until late disease)
- Signal: Confluent T2/FLAIR hyperintensity; confluent sheet-like pattern
- Pathognomonic patterns:
- "Tigroid" pattern: Alternating stripes of demyelinated (T2 bright) and spared (T2 dark) white matter - sparing of perivascular white matter creates a striped appearance; seen in periventricular WM and centrum semiovale
- "Leopard skin" pattern: Punctate foci of T2 sparing within the demyelinated white matter (seen in centrum semiovale)
- Enhancement: Typically no contrast enhancement, even in active disease - important differentiator from X-ALD
- Progression: As disease worsens, inner subcortical WM is eventually involved; corpus callosum, internal capsule, and corticospinal tracts affected
Grainger & Allison: "The tigroid pattern emerges as severe disease develops, with the sheet of white matter signal-intensity abnormality involving the inner half of the subcortical white matter."
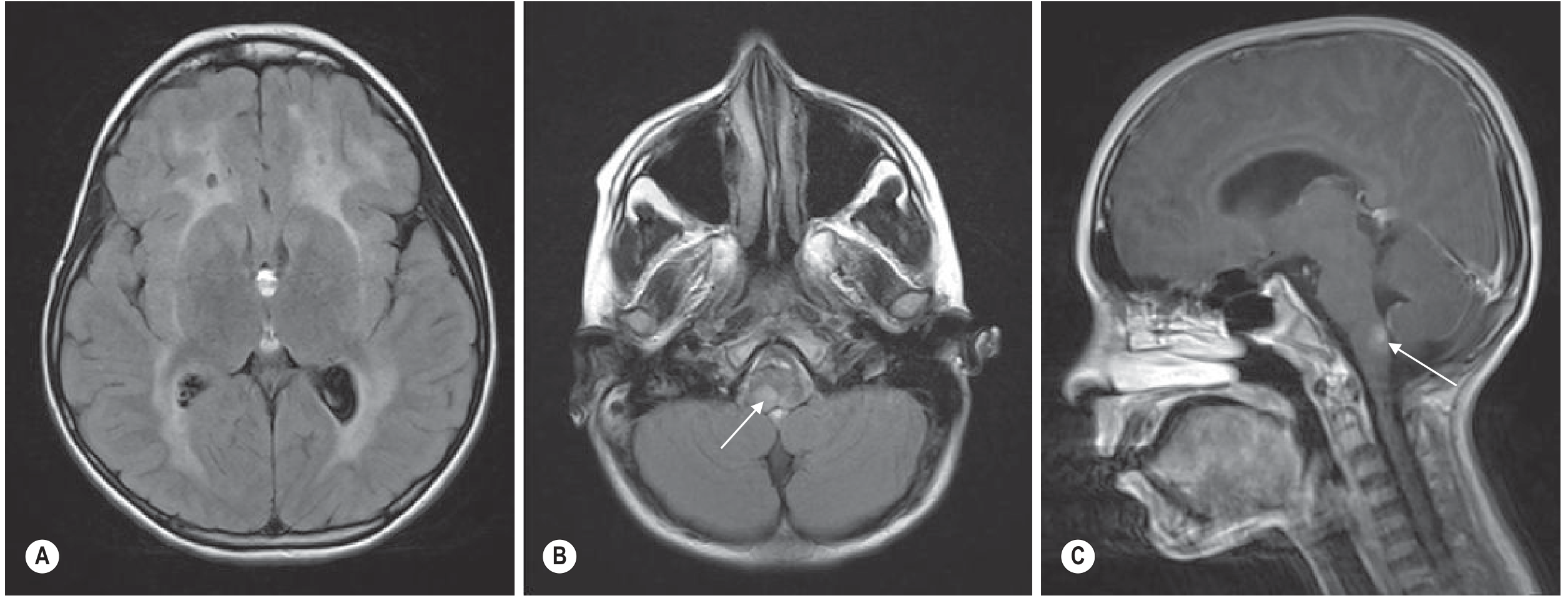
3. Krabbe Disease (Globoid Cell Leukodystrophy)
- Location: Posterior > anterior; starts centrally; specific involvement of:
- Corticospinal tracts as they course through internal capsule and brainstem
- Cerebellar white matter (but dentate nuclei spared)
- Basal ganglia and thalami - characteristically dark thalami on T2 (T2 hypointensity due to iron/mineralization in globoid cells)
- Optic nerves - enlarged/T2 bright in infantile form
- Cervical spinal cord - enlargement in infantile form
- Signal: T2 hyperintensity in white matter; T2 hypointensity in thalami is highly specific
- Enhancement: Can be present in early/active phases
- Infantile form: Very early and severe; optic nerve + cervical cord enlargement is classic
- Adult form: Corticospinal tract hyperintensity prominent
Grainger & Allison: "White matter changes more severely posteriorly and centrally; basal ganglia and thalamic involvement, specifically dark signal in the thalamus on T2 weighted images; cerebellar white matter abnormality, sparing the dentate nuclei; and involvement of the pyramidal tracts within the brainstem."
4. Alexander Disease
- Location: Frontal predominant (anterior > posterior) - this is the key differentiator
- Leukoencephalopathy pattern (van der Knaap criteria - 4 of 5 must be present):
- Extensive cerebral white matter change with frontal predominance
- Rim of T1 hyperintensity / T2 hypointensity around the frontal horns (periventricular rim)
- Abnormality of basal ganglia and thalami
- Brainstem abnormalities (midbrain, medullary involvement)
- Contrast enhancement of one or more of: ventricular lining, frontal WM, basal ganglia, brainstem structures
- Macrocephaly: Almost universal in infantile (Type I) - key clinical-imaging correlation
- Infantile (Type I): Frontal white matter swelling + T2 hyperintensity; basal ganglia and thalami involved; periventricular rim; enhancement
- Juvenile/Adult (Type II): Dorsal medulla + upper cervical cord involvement with enhancement is highly characteristic; less frontal predominance; may have cervicomedullary atrophy
- Rosenthal fibers accumulate in periventricular, perivascular, and subpial locations - the histologic hallmark reflected in the periventricular rim sign
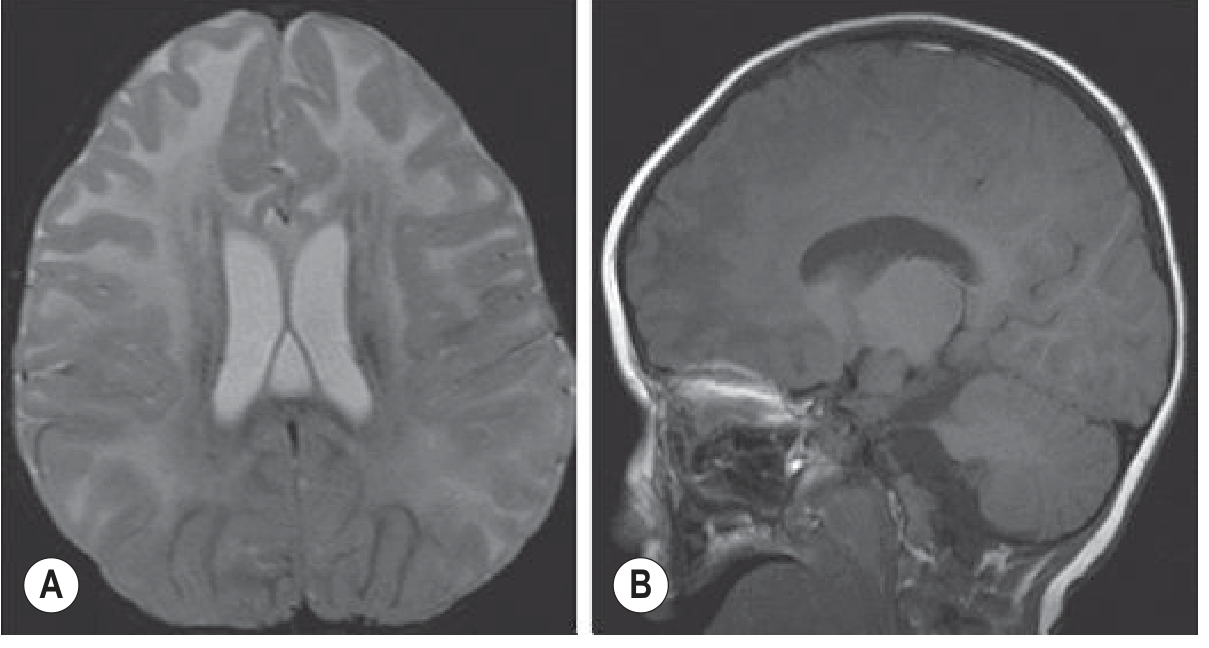
5. Canavan Disease
- Location: Subcortical U-fibers involved early (unlike most leukodystrophies); globus pallidus and thalami involved; cerebellar white matter affected
- Signal: Diffuse T2 hyperintensity throughout white matter including subcortical U-fibers; T2 increase in globus pallidus
- Macrocephaly: Present (due to NAA accumulation causing osmotic brain swelling)
- Key MRS finding: Markedly elevated NAA peak (N-acetylaspartate) at 2.0 ppm - pathognomonic; due to aspartoacylase deficiency
- Diffusion: Restricted diffusion in white matter in early disease (cytotoxic component)
- Late stage: Diffuse cerebral atrophy
Memory hook: Canavan = Canavan → Cortical U-fibers + Cerebellum + Corpus callosum + elevated NAA on MRS
6. Pelizaeus-Merzbacher Disease (PMD)
- Pattern: Hypomyelination (not demyelination) - this is a key distinction
- MRI appearance: The white matter never properly myelinates - it has the appearance of a newborn/young infant brain regardless of actual age; T2 signal in white matter does not suppress (remains bright like unmyelinated WM); T1 WM remains hypointense
- Distribution: Diffuse; cerebellar and brainstem WM also affected; no clear focal pattern
- No enhancement (no inflammatory component)
- Thinned corpus callosum common
- Key distinguishing point: In demyelinating leukodystrophies, there is initial normal myelination followed by loss. In PMD, myelination is arrested from the start.
7. Vanishing White Matter Disease (VWM)
- Hallmark: White matter eventually becomes isointense with CSF on all sequences (literally vanishes)
- Signal: T2/FLAIR hyperintensity in white matter; as disease progresses the signal follows CSF (T1 dark, T2 bright, FLAIR suppressed)
- Cystic change: WM undergoes vacuolation and cavitation - rarefied/cystic appearance
- Distribution: Diffuse cerebral white matter; relative sparing of subcortical U-fibers and posterior fossa (early)
- Trigger: Episodes of acute neurological deterioration follow febrile illness or head trauma - radiologically, acute worsening of white matter changes is seen after stress
- MRS: Absent NAA and Cho peaks over white matter (no viable tissue); lactate/lipid peaks in cystic areas
The Radiology Differential: Quick Comparison Table (High-Yield for PG)
| Feature | X-ALD | MLD | Krabbe | Alexander | Canavan | PMD | VWM |
|---|---|---|---|---|---|---|---|
| Distribution | Posterior→anterior | Central periventricular | Posterior + central | Frontal predominant | U-fibers + pallidi | Diffuse hypomyelination | Diffuse→cystic |
| Corpus callosum | Splenium early | Splenium involved | Involved | Involved | Diffuse | Thin | Involved |
| U-fibers | Spared early | Spared early | Spared early | Involved later | Involved early | Diffuse | Spared early |
| Enhancement | Yes - leading edge | No | Occasionally | Yes - rim/frontal | No | No | No |
| Thalami/BG | Spared | Spared | T2 dark thalami | Involved | Globus pallidus | Spared | Spared |
| Special sign | 3-zone, Loes score | Tigroid/leopard skin | Dark thalami | Periventricular rim, frontal | Elevated NAA (MRS) | Arrested myelination | WM→CSF signal |
| Macrocephaly | No | No | No | Yes | Yes | No | No |
| Gene | ABCD1 (X-linked) | ARSA (AR) | GALC (AR) | GFAP (AD) | ASPA (AR) | PLP1 (X-linked) | EIF2B (AR) |
MRI Protocol for Suspected Leukodystrophy
A complete MRI workup should include:
- T1 axial + sagittal - assess myelination pattern; corpus callosum
- T2 axial - white matter signal; distribution of lesions
- FLAIR - periventricular lesion conspicuity; cystic vs. solid
- DWI/ADC - active demyelination often shows restricted diffusion at the leading edge; cystic WM shows facilitated diffusion
- Post-gadolinium T1 - enhancement pattern (critical for X-ALD staging; Alexander disease)
- MR Spectroscopy - NAA (Canavan ↑↑↑), Cho/NAA ratio, lactate (mitochondrial), absent peaks (VWM)
- Spinal cord MRI if clinically indicated (X-ALD/AMN, Krabbe infantile - cord enlargement)
Radiology PG Mnemonics
- "Posterior to Anterior" = X-ALD (Parieto-occipital first, then advances forward)
- "Frontal = Alexander" (F for Frontal = F for GFAP/Alexander)
- "Tigroid = MLD" (the only leukodystrophy with this striped pattern, and no enhancement)
- "Dark thalami = Krabbe" (T2 hypointense thalami due to globoid cell accumulation)
- "Macrocephaly + elevated NAA = Canavan"
- "Arrested myelination (not demyelination) = PMD"
- "WM becomes CSF = VWM"
- "Enhancement at leading edge = always think X-ALD first"
Sources
- Grainger & Allison's Diagnostic Radiology: Chapter 76, Inborn Metabolic Brain Disorders
- Adams and Victor's Principles of Neurology, 12th Edition
- Goldman-Cecil Medicine
- Leukodystrophy Imaging review, PMC 2024 (Thakkar et al., 2024 - 22 citations)
This is a shared conversation. Sign in to Orris to start your own chat.