Long qt syndrome
Reading File
search_medical_text
Long QT syndrome etiology pathophysiology genetics classification
search_medical_text
Long QT syndrome diagnosis ECG criteria management treatment
search_medical_image
Long QT syndrome ECG findings torsades de pointes
GradeResponse
GradeResponse
GradeResponse
Long QT syndrome (LQTS) is a disorder of delayed ventricular repolarization that increases risk of torsades de pointes and sudden cardiac death.
Quick overview
- ECG hallmark: prolonged corrected QT interval (QTc)
- Typical thresholds: QTc >440 ms in men, >460 ms in women (Harrison’s, p. 7107)
- Can be congenital (ion channel mutations) or acquired (most often medications/electrolyte abnormalities)
Causes
1) Congenital LQTS
- Due to mutations in cardiac ion channel genes (Harrison’s, p. 7107)
- Most common subtypes:
- LQT1 and LQT2: potassium channel related
- LQT3: sodium channel related
These account for most cases (Harrison’s, p. 7107)
Trigger patterns by subtype (classic exam pattern):
- LQT1: exertion, especially swimming
- LQT2: sudden noise/emotional stress
- LQT3: events more often at rest or sleep
(Harrison’s, p. 7107)
2) Acquired LQTS
Common contributors (standard clinical knowledge):
- QT-prolonging drugs (antiarrhythmics, some antibiotics, antipsychotics, antidepressants, etc.)
- Hypokalemia, hypomagnesemia, hypocalcemia
- Bradycardia, structural heart disease, starvation, endocrine issues
Symptoms / presentation
- May be asymptomatic
- Palpitations, presyncope/syncope
- Seizure-like episodes from cerebral hypoperfusion
- Sudden cardiac arrest from torsades de pointes
(Harrison’s congenital presentation often in childhood with syncope/cardiac arrest, p. 7107)
ECG features
- Prolonged QT/QTc
- T-wave abnormalities and sometimes prominent U waves
- Polymorphic VT (torsades) can be triggered by a PVC after prolonged repolarization
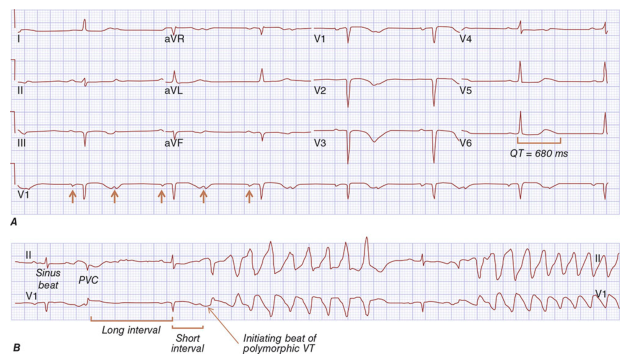
Illustration of prolonged QT and initiation of polymorphic VT/torsades (Harrison’s, p. 7104).
Diagnosis
- Confirm QTc prolongation on quality 12-lead ECG (use Bazett/Fridericia cautiously by HR)
- Review:
- Medication list
- Electrolytes
- Family history of sudden death/syncope
- Consider Schwartz score and genetic testing when congenital LQTS is suspected
- In neonates at risk, ECG timing matters to avoid false overdiagnosis in first days of life; guidelines note repeat assessment and early genetic screening in affected families (Management of CVD in Pregnancy, p. 36)
Management
Acute torsades (emergency)
- Immediate rhythm assessment/defibrillation if unstable
- IV magnesium sulfate
- Correct K/Mg/Ca
- Stop offending QT-prolonging drugs
- Increase heart rate (overdrive pacing/isoproterenol in selected acquired cases)
Chronic / prevention
- Avoid QT-prolonging medications and correct electrolytes
- Beta-blockers are first-line in congenital LQTS (especially LQT1/LQT2)
- Consider ICD for high-risk patients (prior arrest, recurrent events despite therapy)
- Left cardiac sympathetic denervation in selected refractory patients
Practical takeaway
If someone has syncope plus prolonged QTc, treat it as potentially dangerous until proven otherwise, aggressively remove reversible causes, and risk-stratify for congenital LQTS.
If you want, I can also give:
- a QT-prolonging drug red-flag list, or
- a step-by-step ER algorithm for torsades/LQTS.
This is a shared conversation. Sign in to Orris to start your own chat.