Renin angiotensin aldosterone mechanism as pedagogy
Reading File
Finding Sources
Finding Sources
Searching the Web
renin angiotensin aldosterone system RAAS mechanism physiology teaching
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a thorough, pedagogically structured answer. Here it is:
The Renin-Angiotensin-Aldosterone System (RAAS) - A Complete Pedagogical Guide
Overview: What is the RAAS?
The RAAS is a hormonal cascade that provides long-term regulation of arterial blood pressure (Pa), extracellular fluid (ECF) volume, and electrolyte homeostasis. Unlike the baroreceptor reflex (seconds), the RAAS operates over hours to days because it is hormonally - not neurally - mediated. As Guyton and Hall emphasize, the RAAS-kidney interaction is so powerful that "arterial pressure barely changes" across a 10-15x variation in daily salt intake when the system is fully operative. - Guyton and Hall Textbook of Medical Physiology
The Three-Organ Chain: Kidney → Lung/Kidney → Adrenal
The cascade links three anatomical sites. Teaching this geographically helps students remember it:
| Step | Organ | What happens |
|---|---|---|
| 1 | Kidney (JGA) | Renin secreted |
| 2 | Liver | Angiotensinogen is substrate |
| 3 | Lung/Kidney | ACE cleaves Ang I → Ang II |
| 4 | Adrenal cortex | Ang II triggers aldosterone release |
| 5 | Kidney tubules | Aldosterone acts on principal cells |
Step-by-Step Mechanism
Step 1 - The Trigger (What activates renin?)
Renin is stored in juxtaglomerular (JG) cells of afferent arterioles and released by four principal stimuli:
- Decreased renal perfusion pressure - sensed by stretch receptors in afferent arterioles (macula densa pathway)
- Decreased NaCl delivery to the macula densa of the distal convoluted tubule
- Sympathetic stimulation via β₁-adrenergic receptors on JG cells (e.g., hemorrhage, upright posture)
- Decreased plasma Na⁺ (rare - needs ~20 mEq/L drop)
Prorenin is constitutively produced; the active form is released by these stimuli. - Costanzo Physiology 7th Edition
Step 2 - Angiotensinogen → Angiotensin I
Renin is a protease enzyme (not a hormone). It cleaves angiotensinogen - an α-2 globulin made in the liver - by removing the N-terminal 10 amino acids to produce Angiotensin I (a decapeptide).
Angiotensin I has no meaningful biological activity - it is purely a precursor. - Tietz Textbook of Laboratory Medicine, 7th Edition
The amino acid chain at this stage:
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu
Step 3 - Angiotensin I → Angiotensin II (via ACE)
Angiotensin-converting enzyme (ACE), located predominantly in the lungs (also kidneys), cleaves the two C-terminal residues (His-Leu) from Angiotensin I to produce Angiotensin II - an octapeptide and the principal effector molecule of the RAAS.
Here is the peptide conversion pathway from Tietz Textbook of Laboratory Medicine:
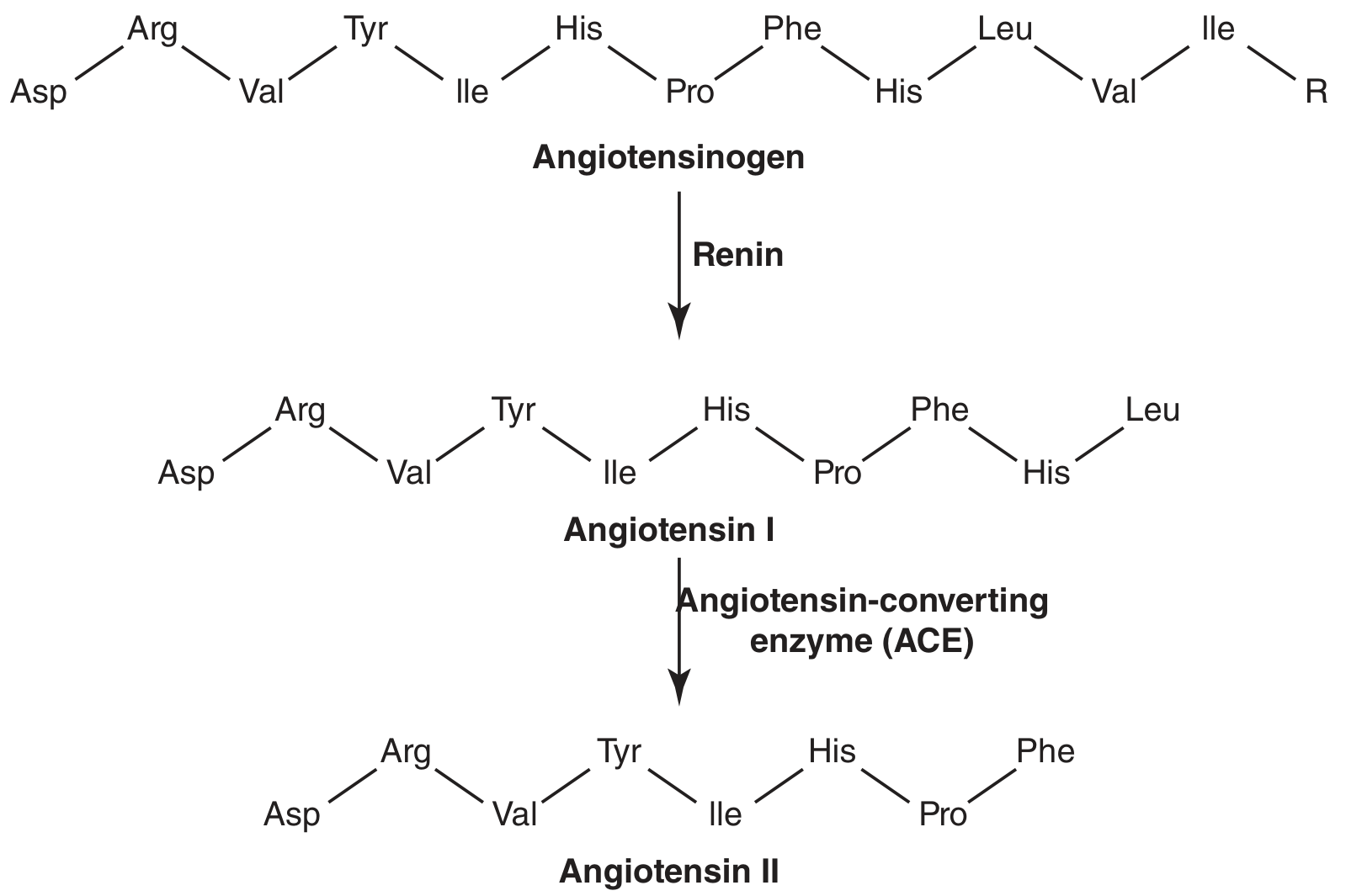
Drug target: ACE inhibitors (captopril, enalapril) block this conversion step, eliminating all downstream Ang II effects.
Step 4 - Angiotensin II: The Master Effector
Angiotensin II acts at AT₁ receptors (Gq-coupled) in four key targets:
A. Adrenal Cortex (Zona Glomerulosa)
- Stimulates synthesis and secretion of aldosterone
- Requires gene transcription and new protein synthesis → hours to days for full effect
B. Kidney (Proximal Tubule)
- Directly stimulates Na⁺-H⁺ exchange, increasing Na⁺ and HCO₃⁻ reabsorption
- This is independent of aldosterone - a separate, faster renal effect
C. Hypothalamus / CNS
- Increases thirst and water intake
- Stimulates ADH (vasopressin) secretion → water reabsorption in collecting ducts
D. Arterioles
- Direct vasoconstriction via IP₃/Ca²⁺ second messenger → ↑ Total Peripheral Resistance (TPR) → ↑ Pa
The Costanzo flowchart captures all four arms beautifully:
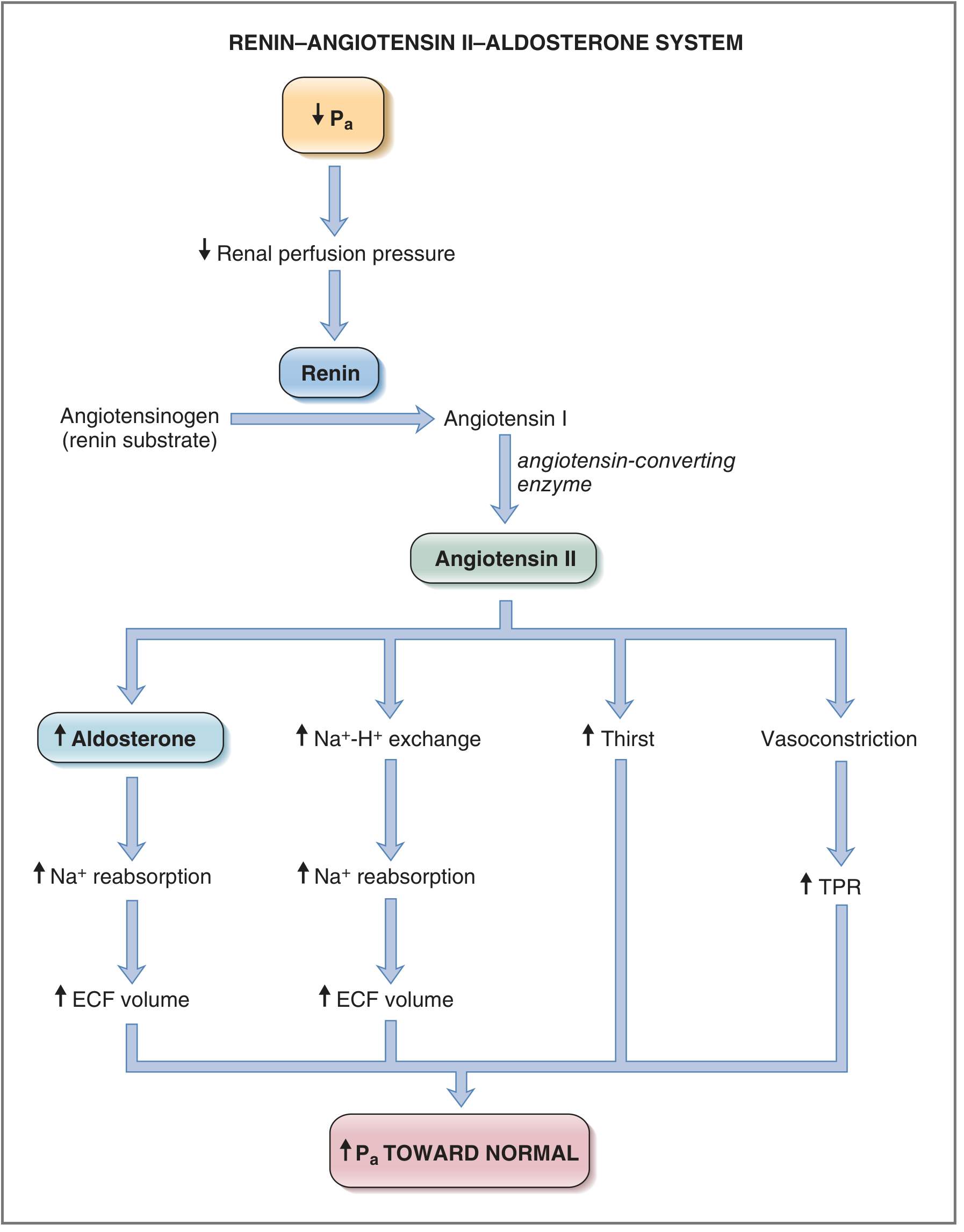
Fig. 4.33 from Costanzo Physiology 7th Edition - the system described in terms of the response to a decrease in Pa. ECF = extracellular fluid; TPR = total peripheral resistance.
Step 5 - Aldosterone: The Final Effector
Aldosterone is a mineralocorticoid steroid from the zona glomerulosa of the adrenal cortex. It acts on principal cells of the distal tubule and collecting duct:
- Upregulates ENaC (epithelial Na⁺ channels on apical membrane) - increases Na⁺ entry
- Upregulates Na⁺/K⁺-ATPase on basolateral membrane - pumps Na⁺ out into blood
- K⁺ and H⁺ are secreted in exchange (explains hypokalemia and alkalosis in hyperaldosteronism)
- Net effect: Na⁺ and water retention → ↑ ECF volume → ↑ blood volume → ↑ Pa
The mechanism requires new protein synthesis, which accounts for the slow time course of this arm. - Costanzo Physiology 7th Edition
Other stimuli for aldosterone release (not via renin):
- Elevated plasma K⁺ (very sensitive - only 1 mEq/L increase is enough)
- ACTH (minor, permissive role)
- These two stimuli are important: they explain why aldosterone can be regulated separately from the RAAS axis. - Ganong's Review of Medical Physiology, 26th Edition
The Negative Feedback Loop
The Ganong feedback diagram:
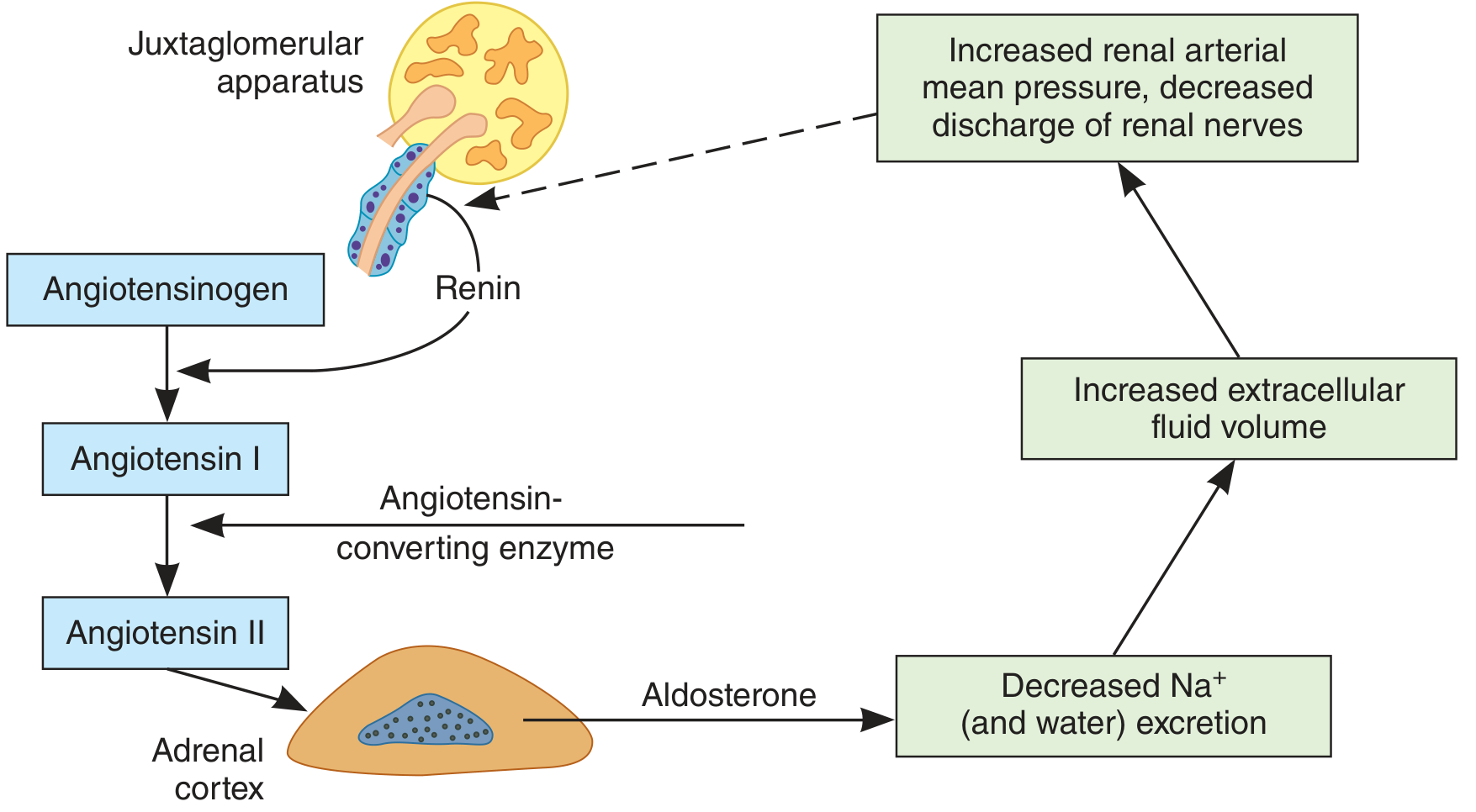
Figure 19-22 from Ganong's Review of Medical Physiology - feedback mechanism regulating aldosterone secretion. The dashed arrow indicates inhibition.
The feedback loop:
- ↓ ECF volume → ↑ Renin → ↑ Ang II → ↑ Aldosterone
- Aldosterone → ↓ Na⁺/water excretion → ↑ ECF volume
- ↑ ECF volume → ↑ renal arterial pressure + ↓ renal nerve discharge → inhibits renin secretion (negative feedback, dashed line)
There is also a proposed "short-loop" feedback: Ang II may directly inhibit renin secretion via AT₁ receptors on JG cells, though this is not conclusively proven. - Tietz Textbook of Laboratory Medicine
Pedagogical Summary Table
| Component | Source | Role | Drug target |
|---|---|---|---|
| Renin | Kidney JGA | Cleaves angiotensinogen | β-blockers ↓ renin; direct renin inhibitors (aliskiren) |
| Angiotensinogen | Liver | Substrate | - |
| Angiotensin I | Plasma | Inactive precursor | - |
| ACE | Lungs/Kidneys | Converts Ang I → Ang II | ACE inhibitors (captopril, enalapril) |
| Angiotensin II | Plasma/Tissues | Vasoconstriction, thirst, ADH, aldosterone | ARBs (losartan, valsartan) |
| Aldosterone | Adrenal zona glomerulosa | Na⁺/water retention, K⁺ excretion | Spironolactone, eplerenone (MR antagonists) |
Clinical Applications (Pedagogical Anchors)
1. Renal artery stenosis - The ischemic kidney hypersecrets renin → Ang II drives up BP. Plasma renin activity is elevated, and renin levels are much higher in the affected renal vein. Treatment: ACE inhibitor (captopril). - Costanzo Physiology 7th Edition, Box 4.2
2. Heart failure - Reduced cardiac output → ↓ renal perfusion → RAAS activation → Na⁺/water retention → worsening preload and afterload. ACE inhibitors and ARBs are cornerstone therapies precisely because they break this vicious cycle.
3. Primary hyperaldosteronism (Conn syndrome) - Aldosterone-secreting adrenal adenoma produces aldosterone independently of renin. The exact mechanism by which adrenal tumors acquire autonomy from the RAAS is not completely understood. Clinically: hypertension + hypokalemia + suppressed renin. - Tietz Textbook of Laboratory Medicine
4. Dietary sodium restriction - Reduces ECF volume → reflexive increase in renal nerve activity → ↑ Renin (before any fall in BP). Salt depletion also upregulates AT₁ receptors in the adrenal cortex and downregulates them in blood vessels, amplifying the aldosterone response but blunting vasoconstriction. - Ganong's Review of Medical Physiology
5. Drug interactions - ACE inhibitors also block bradykinin breakdown (bradykinin is another ACE substrate), causing the well-known ACE inhibitor cough. ARBs do not block ACE, so they don't cause this side effect.
RAAS vs. Baroreceptor Reflex - Teaching Contrast
| Feature | Baroreceptor Reflex | RAAS |
|---|---|---|
| Speed | Seconds | Hours to days |
| Mechanism | Neural | Hormonal |
| Primary effector | Heart rate, TPR | Na⁺ retention, ECF volume |
| Restoration | Partial | Near-complete (nearly infinite feedback gain for long-term Pa) |
| Dysregulation | Minimal long-term impact | Hypertension, HF, CKD |
Teaching Video
This animated RAAS overview from Alila Medical Media is excellent for visual learners (1 million+ views):
Sources: Costanzo Physiology 7th Edition | Guyton and Hall Textbook of Medical Physiology | Ganong's Review of Medical Physiology, 26th Edition | Tietz Textbook of Laboratory Medicine, 7th Edition | StatPearls - Physiology, Renin Angiotensin System
This is a shared conversation. Sign in to Orris to start your own chat.