What is super refectory status epilepticus
"super-refractory status epilepticus" management
super refractory status epilepticus definition causes treatment 2024
Super-Refractory Status Epilepticus (SRSE)
Definition
| Stage | Definition |
|---|---|
| Status Epilepticus (SE) | Continuous seizure activity ≥5 min (convulsive) or ≥10 min (non-convulsive), OR recurrent seizures without return to baseline |
| Refractory SE (RSE) | SE that fails to respond to first-line benzodiazepines AND a second-line IV antiseizure drug |
| Super-Refractory SE (SRSE) | SE persisting or recurring after ≥24 hours of IV anesthetic agents, including recurrence upon anaesthetic withdrawal |
Causes
- Autoimmune/inflammatory encephalitis - anti-NMDA receptor antibodies, anti-GABA-A receptor antibodies; this is an increasingly recognized cause
- Febrile infection-related epilepsy syndrome (FIRES) - in children; the adult equivalent is New-Onset Refractory Status Epilepticus (NORSE)
- Acute structural brain lesions - stroke, TBI, tumor, CNS infection
- Metabolic derangements - severe hyponatremia, hypoglycemia, hepatic encephalopathy
- Toxic/drug causes - drug withdrawal (barbiturates, benzodiazepines), toxin exposure
- Pre-existing epilepsy with subtherapeutic drug levels or acute provocation
- Malignancy (paraneoplastic)
- Cryptogenic - no cause identified in a significant proportion
Pathophysiology
Monitoring
- To detect non-convulsive SE that may persist after motor signs stop
- To guide titration of anesthetic agents - the target endpoint is typically burst suppression on EEG
Treatment
1. IV Anesthetic Agents (Mainstay)
- Midazolam - loading dose 0.2 mg/kg, then infusion 0.1-0.4 mg/kg/h (can escalate significantly); first-line anesthetic
- Propofol - 2 mg/kg bolus then 2-8 mg/kg/h IV drip; effective but risk of propofol infusion syndrome (hypertriglyceridemia, metabolic acidosis, cardiac failure) with prolonged use
- Pentobarbital - 10 mg/kg/h; a reliable agent but causes significant hemodynamic compromise
- Ketamine - increasingly used; NMDA receptor antagonism offers a distinct mechanism and may help when GABA-ergic agents fail
2. Additional Anticonvulsants
3. Ketogenic Diet
4. Immunotherapy (when autoimmune cause suspected or confirmed)
- First-line: IV corticosteroids, IV immunoglobulins (IVIG), plasma exchange (plasmapheresis)
- Second-line: Rituximab (375 mg/m² weekly x4 weeks), cyclophosphamide
- These are often started empirically in SRSE while antibody testing is pending, as delay worsens outcome in autoimmune encephalitis
5. Neurostimulation (selected cases)
- Electroconvulsive therapy (ECT) - paradoxically, ECT can terminate prolonged SE
- Vagus nerve stimulation (VNS)
- Thalamic deep brain stimulation - case reports of success
6. Surgical/Interventional
- Resective epilepsy surgery - if a discrete structural lesion is the source
- Applicable only in highly selected patients
Key Related Syndrome: NORSE / FIRES
Prognosis
- Mortality ~3% (convulsive form), higher in older patients and those with structural or anoxic causes
- Survivors often have new epilepsy, cognitive deficits, behavioral changes, or need for long-term institutionalization
- FIRES/NORSE has especially poor neurological outcomes despite seizure cessation
What are the monoclonal antibodies used in the t cell all and chemotherapy regimen
monoclonal antibodies T-cell ALL treatment regimen 2024 2025
https://pmc.ncbi.nlm.nih.gov/articles/PMC12712861
daratumumab brentuximab alemtuzumab T-cell ALL monoclonal antibody CD52 CD38 CD7 CAR-T 2024
Monoclonal Antibodies in T-Cell ALL and Chemotherapy Regimens
The Key Challenge with T-Cell ALL
Standard Chemotherapy Backbone for T-ALL
| Regimen | Components |
|---|---|
| Hyper-CVAD (US-preferred) | Hyperfractionated cyclophosphamide, vincristine, doxorubicin (Adriamycin), dexamethasone - alternating with HD methotrexate + cytarabine |
| BFM (Berlin-Frankfurt-Münster) | European standard; induction + consolidation + maintenance |
- Nelarabine (ara-G prodrug) - the ONLY FDA-approved agent specifically for T-ALL (approved 2005); a purine nucleoside analogue with selective T-lymphoblast toxicity
- Pegylated asparaginase (PEG-ASP) - depletes asparagine, starving leukemic cells; now standard in most frontline regimens
- High-dose methotrexate - essential for CNS prophylaxis
- High-dose cytarabine - consolidation
- 6-Mercaptopurine + methotrexate - maintenance (2-2.5 years)
Monoclonal Antibodies Used (or Being Investigated) in T-ALL
1. Alemtuzumab (anti-CD52) - Established
- Target: CD52, a glycoprotein highly expressed on both T and B lymphocytes (malignant and normal)
- Type: Humanized IgG1 monoclonal antibody
- Role in T-ALL: Used in salvage/relapsed T-ALL and T-cell prolymphocytic leukemia (T-PLL). Also used as lymphodepletion conditioning before CAR-T cell therapy (e.g., UCART programs)
- Key toxicity: Profound lymphodepletion - requires PCP, CMV, and herpes prophylaxis for months after therapy
- Harrison's lists it under mature T-cell neoplasm responses; Cellular and Molecular Immunology (table 18.1) explicitly lists alemtuzumab against "CLL, CTCL, and T-cell lymphoma"
2. Daratumumab (anti-CD38) - Investigational in T-ALL
- Target: CD38, which is expressed on T-ALL blasts (especially ETP-ALL)
- Type: Human IgG1 monoclonal antibody (well-established in multiple myeloma)
- Role in T-ALL: Being actively investigated - a Blood 2024 review specifically addresses daratumumab's potential in T-cell leukemias, noting its mechanism via complement-dependent cytotoxicity, ADCC, and direct apoptosis
- Not yet standard of care; being explored in clinical trials
3. Brentuximab Vedotin (anti-CD30) - Selective Use
- Target: CD30
- Type: Antibody-drug conjugate (ADC) - anti-CD30 + MMAE (microtubule inhibitor)
- Role: CD30 can be expressed on a subset of T-ALL/T-lymphoblastic lymphoma cases; used in those with confirmed CD30 expression, more commonly in anaplastic large-cell lymphoma (ALCL)
- Flow cytometry is used to assess CD30 status before use (per Tietz)
4. Blinatumomab (anti-CD19/CD3 BiTE) - Primarily B-ALL, but T-ALL context
- Target: Bispecific T-cell engager - bridges CD19+ leukemic cells to CD3+ T cells
- Note: This is a B-ALL drug (not applicable in T-ALL directly), but it is mentioned here because in combination therapies, it is sometimes used when lineage switch or mixed phenotype occurs
- 70-80% MRD conversion rate in B-ALL (Harrison's)
5. CAR-T Cells Targeting T-ALL Antigens (Emerging, not yet mAbs in the classical sense)
- CD7 CAR-T cells - CD7 is expressed on most T-ALL blasts; the challenge is fratricide (T-CAR cells also express CD7), being overcome with CD7 knockout or CD7 blocking strategies
- CD5-targeted CAR-T cells - another T-cell antigen being explored
- These represent the most active frontier for T-ALL immunotherapy
Agents Under Investigation (Not Yet Standard)
| Agent | Target | Mechanism |
|---|---|---|
| Venetoclax | BCL-2 | BH3-mimetic; especially promising in ETP-ALL (high BCL-2 expression) |
| Navitoclax | BCL-2/BCL-XL | BH3-mimetic |
| Bortezomib | Proteasome | Being explored with chemotherapy |
| γ-secretase inhibitors | NOTCH1 | NOTCH1 mutations occur in ~60% of T-ALL |
| HDAC inhibitors (vorinostat, romidepsin) | Histone deacetylases | Epigenetic targeting |
Treatment by T-ALL Subtype (Harrison's 22nd ed., Table 111-4)
| T-ALL Subtype | Treatment | OS |
|---|---|---|
| Early pro/pre-T (ETP-ALL) | Intensive chemo + nelarabine + allogeneic SCT | 40-50% |
| Cortical/Thymic T-ALL | Intensive chemo + nelarabine, no SCT needed | 70-84% |
| Mature T-ALL | Intensive chemo + nelarabine + SCT | ~30-40% |
Summary: Why T-ALL Lags Behind B-ALL in Antibody Therapy
What are the reason for the aptt are high in the b all
prolonged aPTT B-cell ALL causes asparaginase DIC coagulopathy mechanism
Reasons for Prolonged aPTT in B-Cell ALL
1. L-Asparaginase Therapy (Most Common and Important Cause)
| Protein Depleted | Effect on aPTT |
|---|---|
| Fibrinogen | Directly prolongs aPTT (factor I in common pathway) |
| Antithrombin (AT III) | Leads to paradoxical thrombosis (AT depletion removes natural anticoagulation) |
| Factors V, VIII, IX | Key intrinsic/common pathway factors - direct aPTT prolongation |
| Protein C and Protein S | Anticoagulant proteins - loss promotes thrombosis |
| Plasminogen | Impairs fibrinolysis |
2. Disseminated Intravascular Coagulation (DIC)
- B-ALL blasts can express tissue factor (TF) and release procoagulant granules, triggering uncontrolled thrombin generation
- This leads to consumption of clotting factors (I, II, V, VIII, X, XIII) and platelets
- The Washington Manual lists DIC as a direct oncological emergency complication of ALL
- Results in: prolonged aPTT + prolonged PT + low fibrinogen + low platelets + elevated D-dimers
3. Hepatic Dysfunction / Liver Infiltration
- The liver is the site of synthesis for all intrinsic and common pathway clotting factors except vWF (which is endothelial)
- In B-ALL, the liver can be infiltrated by leukemic blasts (hepatomegaly occurs in ~60% of childhood ALL)
- Additionally, chemotherapy-induced hepatotoxicity (especially L-asparaginase, methotrexate) impairs hepatic synthetic function
- Result: reduced production of factors II, V, VIII, IX, X, XI, fibrinogen → prolonged aPTT ± PT
4. Lupus Anticoagulant / Antiphospholipid Antibodies
- Malignancy, including leukemia, can induce acquired antiphospholipid antibodies
- Lupus anticoagulant (LA) prolongs phospholipid-dependent clotting tests including aPTT in vitro, but paradoxically causes thrombosis in vivo
- Dermatology 5e's interpretation table explicitly lists lupus anticoagulant under "Normal PT, prolonged aPTT"
- In B-ALL: B-cell dysregulation and cytokine storm can trigger LA production
5. Acquired Factor Inhibitors
- B-ALL can rarely be associated with development of acquired inhibitors against specific clotting factors (most commonly factor VIII)
- These are autoantibodies that neutralize clotting factor activity
- Result: isolated aPTT prolongation that does not correct on mixing study (unlike factor deficiency which does correct)
6. Heparin Therapy (Iatrogenic)
- Patients with ALL often have central venous catheters (CVCs) flushed with heparin, or receive therapeutic/prophylactic heparin to counteract asparaginase-induced AT deficiency and thrombosis risk
- Heparin directly prolongs aPTT by accelerating antithrombin activity against thrombin and factor Xa
- This is a very common iatrogenic cause in the inpatient setting
7. Vitamin K Deficiency
- ALL patients are frequently on broad-spectrum antibiotics (for infection prophylaxis/treatment), which deplete gut flora that produce vitamin K
- Poor oral intake and malnutrition during induction chemotherapy also reduces vitamin K
- Vitamin K deficiency impairs carboxylation of factors II, VII, IX, X, protein C, protein S
- Primarily elevates PT, but with severe deficiency, aPTT also rises (factors IX and X are shared with the intrinsic pathway)
8. Disease-Related Consumption (Leukemic Coagulopathy)
- The leukemic process itself, even before treatment, causes aPTT/PT prolongation
- Studies in childhood ALL confirm higher PT and aPTT at diagnosis compared to healthy controls, attributed to blast-mediated procoagulant activity and baseline coagulation factor depletion
- This is separate from and additive to treatment-related coagulopathy
Diagnostic Framework: Interpreting the aPTT in B-ALL
| Finding | Likely Cause |
|---|---|
| ↑ aPTT + ↑ PT + ↓ fibrinogen + ↓ platelets | DIC |
| ↑ aPTT + ↑ PT + normal platelets | L-asparaginase hepatotoxicity / liver infiltration |
| ↑ aPTT + normal PT | Heparin, lupus anticoagulant, acquired factor VIII inhibitor, isolated factor IX/XI/XII deficiency |
| aPTT corrects on mixing study | Factor deficiency (asparaginase-related depletion) |
| aPTT does NOT correct on mixing study | Inhibitor (lupus anticoagulant, acquired factor VIII Ab) |
What are the reason to cause the referactory seizures status epilepticus
refractory status epilepticus causes etiology mechanisms pharmacoresistance 2024
Causes of Refractory Status Epilepticus (RSE)
PART A: Neurobiological Mechanisms That Make SE Refractory
1. GABA-A Receptor Internalization (Most Important Mechanism)
- During prolonged SE, synaptic GABA-A receptors are sequestered and internalized into the cell, away from the synapse
- This removes the drug target for all benzodiazepines (which work by enhancing GABA-A activity)
- Simultaneously, NMDA (excitatory) receptors are upregulated at the synaptic membrane
- The result: inhibitory signaling fails, excitatory signaling amplifies - SE becomes self-perpetuating and benzodiazepine-resistant
- This is why time to treatment is critical - the longer SE continues, the more irreversible these receptor changes become
2. Excitotoxic Cascade
- Sustained neuronal firing leads to excess intracellular calcium entry via NMDA receptors
- This triggers mitochondrial failure, free radical production, and neuronal apoptosis
- As more neurons are damaged, normal inhibitory networks break down further
3. Drug Efflux Transporter Overexpression
- P-glycoprotein and other multidrug resistance (MDR) transporters are upregulated at the blood-brain barrier during SE
- These pumps actively expel antiseizure medications back out of the brain
- This reduces effective drug concentrations at the epileptic focus despite adequate systemic levels
4. Neuroinflammation
- SE itself triggers microglial activation and release of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6)
- Neuroinflammation lowers seizure threshold and promotes further seizure activity
- Creates a vicious cycle: SE → neuroinflammation → more SE
PART B: Clinical Causes (Underlying Etiologies)
1. Autoimmune / Inflammatory Encephalitis (Most Common Cause of New-Onset RSE)
| Antibody / Syndrome | Target | Seizure Features |
|---|---|---|
| Anti-NMDA receptor encephalitis | GluN1 subunit | Very refractory SE, often with psychiatric prodrome |
| Anti-GABA-A receptor encephalitis | GABA-A receptor | Prominent SE, often requires anesthetic coma; MRI shows multifocal FLAIR changes |
| Anti-GABA-B receptor encephalitis | GABA-B receptor | SE + cognitive changes |
| Anti-LGI1 encephalitis | Voltage-gated K+ channel complex | Faciobrachial dystonic seizures, then SE |
| Rasmussen encephalitis | Uncertain/GluR3 | Progressive hemispheric destruction, epilepsia partialis continua |
| ADEM | Myelin | SE in children post-infection |
| Paraneoplastic (anti-Hu, anti-Yo, etc.) | Intranuclear antigens | Associated with occult malignancy |
| FIRES / NORSE | Unknown (often cryptogenic) | Febrile illness → refractory SE in children/adults |
2. CNS Infections
- Viral encephalitis: Herpes simplex virus (HSV) is the most important - has strong predilection for temporal lobes
- Bacterial meningitis: Purulent meningitis can cause cortical irritation and SE
- Tuberculous meningitis / encephalitis
- Cerebral abscess
- Neurocysticercosis (most common infectious cause of SE worldwide in endemic regions)
- COVID-19 encephalopathy (increasingly recognized)
- Fungal meningitis in immunocompromised patients
3. Cerebrovascular Disease
- Acute ischemic stroke (particularly cortical and large territory strokes)
- Intracerebral hemorrhage (cortical irritation from blood)
- Subarachnoid hemorrhage
- Cerebral venous thrombosis (CVT) - classically causes refractory SE, especially in women
- Posterior Reversible Encephalopathy Syndrome (PRES) - hypertension/eclampsia-related
- Cavernous malformations / arteriovenous malformations
4. Metabolic Derangements
| Metabolic Cause | Mechanism |
|---|---|
| Hyponatremia | Cerebral edema, reduced seizure threshold |
| Hypoglycemia | Neuronal energy failure |
| Hyperglycemia (non-ketotic) | Paradoxically lowers GABA activity |
| Hypocalcemia | Increases neuronal excitability (Ca²+ stabilizes membranes) |
| Hypomagnesemia | Removes NMDA receptor block (Mg²+ normally blocks NMDA) |
| Hyperammonemia | Liver failure / urea cycle disorders |
| Acidosis / uremia | Renal failure, toxic metabolite accumulation |
| Wernicke encephalopathy | Thiamine deficiency - especially in alcoholics |
| Pyridoxine (B6) deficiency | Neonates; B6 is a cofactor for GABA synthesis |
5. Drugs and Toxins
- Withdrawal states: Alcohol, benzodiazepines, barbiturates - GABAergic withdrawal causes hyperexcitability and notoriously refractory SE
- Subtherapeutic antiseizure drug levels (non-compliance, drug interaction, nil-by-mouth in known epileptics)
- Isoniazid (INH): Depletes pyridoxine → impairs GABA synthesis → GABA-independent seizures that are resistant to standard drugs
- Cocaine, amphetamines, synthetic cannabinoids, bath salts
- Baclofen withdrawal
- Tramadol, tacrolimus, alkylating agents (chemotherapy)
- Local anesthetic toxicity (bupivacaine)
6. Structural / Traumatic Brain Lesions
- Head trauma: Epidural/subdural/subarachnoid hemorrhage, diffuse axonal injury
- Intracranial tumors: Gliomas, meningiomas, metastases, lymphoma, gangliogliomas
- Focal cortical dysplasia (developmental malformation) - inherently epileptogenic
- Hippocampal sclerosis - mesial temporal sclerosis often causes pharmacoresistant temporal lobe SE
7. Hypoxic-Ischemic Brain Injury
- Post-cardiac arrest: The most common cause of SE in ICU patients
- Global anoxia leads to widespread excitotoxicity
- Post-anoxic myoclonic SE carries very poor prognosis
8. Genetic / Hereditary Conditions
- Tuberous sclerosis complex (TSC): Multiple cortical tubers are epileptogenic
- Dravet syndrome (SCN1A mutation): Temperature-sensitive SE, notoriously drug-resistant
- CDKL5 deficiency, KCNQ2 mutations
- Mitochondrial diseases (MERRF, MELAS): Energy failure in neurons
- Porphyria: Acute attacks can cause refractory SE
- Wilson disease: Copper deposition in basal ganglia and cortex
9. Special Entities: NORSE and FIRES
- NORSE (New-Onset Refractory SE): RSE in a patient with no prior epilepsy, no acute toxic/metabolic cause, and no clear structural cause. In adults, autoimmune encephalitis is identified in only ~50% - over half remain cryptogenic
- FIRES (Febrile Infection-Related Epilepsy Syndrome): Pediatric NORSE with a febrile prodrome 24h-2 weeks before SE onset. No cause consistently identified; IL-1β pathway suspected
- Both are characterized by SE that is profoundly resistant to all standard therapies
Summary Classification
| Category | Key Examples |
|---|---|
| Autoimmune | Anti-NMDA, anti-GABA-A, FIRES/NORSE, paraneoplastic |
| Infectious | HSV encephalitis, bacterial meningitis, neurocysticercosis |
| Cerebrovascular | Stroke, CVT, hemorrhage, PRES |
| Metabolic | Hyponatremia, hypoglycemia, hypocalcemia, Wernicke's |
| Toxic/Drug | Alcohol withdrawal, INH, benzodiazepine withdrawal |
| Structural | Tumor, trauma, cortical dysplasia, hippocampal sclerosis |
| Hypoxic-ischemic | Post-cardiac arrest |
| Genetic | Dravet, TSC, mitochondrial diseases |
| Cryptogenic | >50% of NORSE; no cause found despite full workup |
Exgulff prices in the anticancer
drug efflux pumps P-glycoprotein MDR anticancer resistance mechanism ABC transporters 2024
Drug Efflux Pumps in Anticancer Drug Resistance
The Core Mechanism - How Efflux Pumps Work
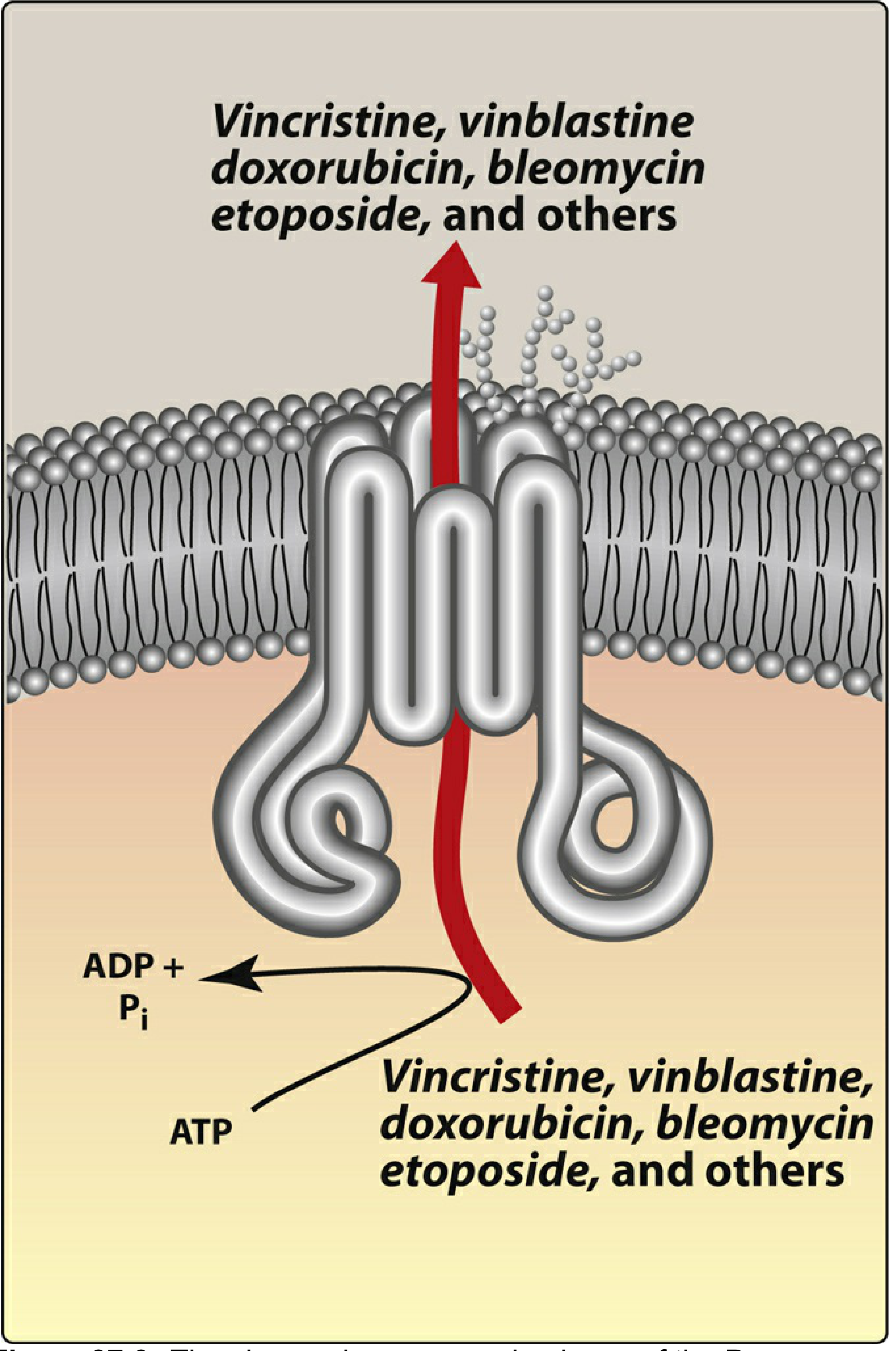
- Recognise hydrophobic drug molecules inside the cell membrane
- Use ATP hydrolysis to power conformational changes
- Expel the drug out of the cell before it can reach its intracellular target
- Result: drug concentration inside the cell stays below cytotoxic levels despite adequate dosing
The Three Major Efflux Pumps in Cancer
1. P-Glycoprotein (P-gp) / MDR1 / ABCB1 - The Most Important
| Feature | Detail |
|---|---|
| Gene | ABCB1 (also called MDR1) |
| Protein | P-glycoprotein ("permeability glycoprotein") |
| Also known as | MDR1, ABCB1, CD243 |
| Structure | Two halves, each with 6 transmembrane domains + ATP-binding domain = 12 transmembrane loops total forming a central channel |
| Substrate selectivity | Broad - primarily hydrophobic, cationic drugs |
| Normal expression | Kidney, liver, pancreas, small intestine, colon, adrenal gland, blood-brain barrier |
- Vinca alkaloids: vincristine, vinblastine, vinorelbine
- Taxanes: paclitaxel, docetaxel
- Anthracyclines: doxorubicin (Adriamycin), daunorubicin, epirubicin
- Epipodophyllotoxins: etoposide, teniposide
- Bleomycin
- Actinomycin D (dactinomycin)
- Imatinib (partially)
2. Multidrug Resistance-Associated Proteins (MRP / ABCC family)
| Transporter | Gene | Key Substrates |
|---|---|---|
| MRP1 | ABCC1 | Doxorubicin, etoposide, methotrexate, vincristine (as glutathione conjugates) |
| MRP2 | ABCC2 | Cisplatin, methotrexate, vinca alkaloids |
| MRP4 | ABCC4 | Methotrexate, 6-mercaptopurine, thiopurines |
3. Breast Cancer Resistance Protein (BCRP / ABCG2)
| Feature | Detail |
|---|---|
| Gene | ABCG2 |
| Normal function | Transports sulfated steroids, uric acid, xenobiotics |
| Named | Found originally in breast cancer cell lines resistant to drugs without P-gp or MRP overexpression |
| Key substrates | Methotrexate, topotecan, irinotecan, imatinib, gefitinib, mitoxantrone |
| Special note | Highly expressed in cancer stem cells - this contributes to "side population" phenotype and intrinsic drug tolerance |
Complete ABC Transporter Classification (Medically Relevant)
| Subfamily | Gene Examples | Clinical Relevance |
|---|---|---|
| ABCB (MDR) | ABCB1 = MDR1/P-gp | Major anticancer drug efflux |
| ABCC (MRP/CFTR) | ABCC1 (MRP1), ABCC2 (MRP2) | Anticancer efflux + CFTR (cystic fibrosis) |
| ABCG | ABCG2 (BCRP) | Breast/lung cancer resistance; stem cells |
Why Intrinsic vs. Acquired MDR?
- Some tumor types naturally express high P-gp at diagnosis - this is why adenocarcinomas (colon, kidney, pancreas, liver cancers) are inherently resistant to many chemotherapy agents. These organs physiologically express P-gp at high levels to protect themselves from toxins.
- Cancer cells that initially respond to chemotherapy can develop resistance through gene amplification of ABCB1 (MDR1), upregulating P-gp expression
- Chromosomal abnormalities consistent with gene amplification have been observed in resistant cells in culture
- Driven by selective pressure - exposure to suboptimal doses selects for drug-resistant clones
Drugs That Inhibit Efflux Pumps (P-gp Inhibitors)
| Generation | Examples | Problem |
|---|---|---|
| 1st generation | Verapamil, cyclosporine, tamoxifen | Unacceptable cardiac/immunosuppressive toxicity at doses needed |
| 2nd generation | Dexverapamil, PSC-833 (valspodar) | Pharmacokinetic interactions; clinical trials failed |
| 3rd generation | Tariquidar, zosuquidar, elacridar | More selective; clinical trials ongoing - not yet approved |
Strategies to Overcome Efflux Pump-Mediated MDR
| Strategy | Examples |
|---|---|
| P-gp inhibitor co-administration | Tariquidar + paclitaxel (clinical trials) |
| Nanoparticle drug delivery | Nanoformulations evade P-gp recognition; albumin-bound paclitaxel (nab-paclitaxel) has some advantage |
| Non-P-gp substrate drugs | Design of new drugs not recognized by ABC transporters |
| Structural modification | Chemical conjugation to produce P-gp inhibitor-drug hybrids |
| MDR gene approach | Transducing hematopoietic stem cells with MDR1 to protect bone marrow during high-dose chemotherapy (Harrison's) |
| Combination chemotherapy | Multiple drugs simultaneously overwhelm the pump capacity |
Clinical Impact Summary
- P-gp overexpression predicts poor response and shorter survival in AML, ALL, breast cancer, ovarian cancer, lymphoma
- Cabazitaxel (a taxane) was specifically developed to be a poor P-gp substrate and is used in docetaxel-resistant prostate cancer
- Measurement of MDR1/P-gp expression on tumor cells is now used in some centers as a predictive biomarker
Why the vanco and ceftrixone combination are used in the meningities
vancomycin ceftriaxone combination bacterial meningitis rationale empirical therapy 2024
Why Vancomycin + Ceftriaxone is Used in Bacterial Meningitis
The Core Problem: Penicillin/Cephalosporin-Resistant Pneumococcus
- S. pneumoniae is the most common cause of community-acquired bacterial meningitis in adults and is associated with the highest mortality (~20-30%)
- Historically, penicillin was the drug of choice - this no longer works reliably
- Widespread resistance to penicillins AND extended resistance to 3rd-generation cephalosporins has emerged globally
- Recent data (Brazil 2023) show cephalosporin resistance in >30% of S. pneumoniae isolates from pediatric meningitis cases
- A cephalosporin-resistant strain treated with ceftriaxone alone = treatment failure and death
What Each Drug Contributes
Ceftriaxone (3rd-Generation Cephalosporin) - The Broad Backbone
| Property | Detail |
|---|---|
| Mechanism | Inhibits bacterial cell wall synthesis by binding penicillin-binding proteins (PBPs) |
| Spectrum | Gram-positive: susceptible S. pneumoniae, group B streptococci; Gram-negative: N. meningitidis, H. influenzae, many Enterobacteriaceae |
| CSF penetration | Excellent - penetrates inflamed blood-brain barrier well (β-lactams enter poorly through normal BBB but penetrate inflamed meninges) |
| Bactericidal | Yes - achieves CSF levels 10-20x the minimum bactericidal concentration (MBC) needed for cure |
| Dosing in meningitis | 2g IV q12h (adult) |
| Key organisms covered | S. pneumoniae (susceptible strains), N. meningitidis, H. influenzae |
Vancomycin (Glycopeptide) - The Resistance Shield
| Property | Detail |
|---|---|
| Mechanism | Inhibits cell wall synthesis by binding D-Ala-D-Ala terminus of peptidoglycan precursors - different mechanism from β-lactams |
| Spectrum | Gram-positive organisms: MRSA, penicillin/cephalosporin-resistant S. pneumoniae |
| CSF penetration | Poor through normal BBB; variable through inflamed meninges (relies on meningeal inflammation for entry) |
| Bactericidal | Yes, but slower than β-lactams |
| Dosing in meningitis | 45-60 mg/kg/day IV divided q8-12h (higher doses required to ensure CSF penetration) |
| Key role | Covers highly resistant S. pneumoniae that would otherwise escape ceftriaxone |
Why the Combination Is Synergistic and Necessary
| Organism | Ceftriaxone alone | Vancomycin alone | Combination |
|---|---|---|---|
| Susceptible S. pneumoniae | ✓ | ✓ | ✓✓ |
| Penicillin/cephalosporin-resistant S. pneumoniae | ✗ FAILS | ✓ (if adequate CSF levels) | ✓✓ SAFE |
| N. meningitidis | ✓✓ | ✗ | ✓✓ |
| H. influenzae | ✓✓ | ✗ | ✓✓ |
| Group B streptococci | ✓ | ✓ | ✓✓ |
The Dexamethasone Complication
- Dexamethasone is given adjunctively (10 mg IV q6h x4 days, starting 20 minutes BEFORE the first antibiotic dose) to reduce neuroinflammation, decrease cytokine-mediated damage, and prevent sensorineural hearing loss
- However, dexamethasone stabilizes and reduces inflammation in the BBB → reduces vancomycin penetration into CSF
- This creates a paradox: the steroid that reduces morbidity also impairs delivery of the very drug needed for resistant pneumococcus
- Increase vancomycin dose to 45-60 mg/kg/day (higher than usual) to compensate for reduced penetration
- Intraventricular vancomycin in selected cases (bypasses BBB entirely)
- A 2007 prospective study (Ricard et al.) reassured clinicians: despite dexamethasone, CSF vancomycin levels remained ≥4x the MIC in all patients, and none had persistent positive cultures. CSF vancomycin was proportional to serum levels, so dose-adjusting using serum levels works.
When to Modify the Regimen
| Clinical Scenario | Add to Vanco + Ceftriaxone |
|---|---|
| Age >55 or immunocompromised | + Ampicillin (covers Listeria monocytogenes - not covered by cephalosporins) |
| Otitis, sinusitis, mastoiditis | + Metronidazole (covers anaerobes from contiguous source) |
| Post-neurosurgery / nosocomial | Replace with vanco + cefepime or meropenem (covers Pseudomonas, MRSA) |
| Penicillin allergy | Vancomycin + chloramphenicol (for Gram-negative coverage), or moxifloxacin |
| Culture confirms susceptible pneumococcus | Discontinue vancomycin - ceftriaxone alone is sufficient |
| Confirmed Listeria | Discontinue ceftriaxone - switch to ampicillin ± gentamicin (cephalosporins are NOT active against Listeria) |
Summary: The One-Line Rationale
Ceftriaxone covers Gram-negatives (meningococcus, H. influenzae) and susceptible pneumococcus; vancomycin covers the penicillin/cephalosporin-resistant pneumococcus that ceftriaxone would miss - together they provide complete empirical cover for all common community-acquired meningitis organisms before culture results are available.
- Ceftriaxone: 2g IV q12h
- Vancomycin: 45-60 mg/kg/day divided q8-12h
Glycoprotein positive
In auto immune encephalitis
glycoprotein positive autoimmune encephalitis MOG CASPR2 LGI1 VGKC antibodies clinical features 2024
Glycoprotein-Positive Autoimmune Encephalitis
1. MOG (Myelin Oligodendrocyte Glycoprotein) - Anti-MOG Antibody Disease (MOGAD)
What is MOG?
Who gets it?
- Predominantly children and young adults
- Both sexes equally affected
- Not typically associated with tumors (rare exception: ovarian teratoma)
Clinical Phenotypes
| Presentation | Features |
|---|---|
| Optic neuritis | Often bilateral and synchronous (distinguishes from MS); painful, severe vision loss |
| Transverse myelitis | Often longitudinally extensive |
| ADEM (Acute Disseminated Encephalomyelitis) | More common in children; multifocal demyelination with encephalopathy |
| Cortical encephalitis | Seizures, confusion, cortical FLAIR changes on MRI |
| Brainstem/cerebellar syndrome | Ataxia, nystagmus, diplopia |
| NMOSD-like (AQP4-seronegative) | NMO phenotype but MOG-positive, AQP4-negative |
Key Distinguishing Features from MS and AQP4-NMOSD:
- Bilateral, simultaneous optic neuritis (MS usually unilateral)
- Good recovery between attacks (unlike AQP4-NMOSD)
- Meninges can be involved (meningitis-like presentation)
- Longitudinally extensive cord lesion with "H-sign" on MRI
MRI:
- Fluffy, poorly marginated lesions (contrast to the sharp black holes of MS)
- Optic nerve enhancement - often extensive and bilateral
- Cortical lesions with leptomeningeal enhancement in cortical encephalitis
Treatment:
- Acute: IV methylprednisolone; IVIG; plasma exchange for severe attacks
- Maintenance: Azathioprine, mycophenolate, or rituximab for relapsing disease
- ~85% respond to immunotherapy; relapses in ~30%
- Important: Do NOT treat with natalizumab or fingolimod (used in MS) - may worsen MOGAD
2. LGI1 (Leucine-Rich Glioma-Inactivated 1) - VGKC Complex Glycoprotein
What is LGI1?
Who gets it?
- Elderly men predominantly (median age 60 years, male > female)
- Typically not cancer-associated (<10% have tumor, usually thymoma or neuroendocrine)
Clinical Features (Classic Triad):
- Faciobrachial Dystonic Seizures (FBDS) - pathognomonic; brief (<3 sec), frequent (up to 100/day) ipsilateral arm and face dystonic jerks; precede full limbic encephalitis
- Limbic encephalitis - subacute memory loss, confusion, temporal lobe seizures
- Hyponatremia (60% of patients) - due to SIADH from hypothalamic LGI1 expression
Additional Features:
- REM sleep behavior disorder
- Psychiatric disturbances (anxiety, depression)
- MRI: medial temporal lobe FLAIR signal (hippocampus, amygdala); sometimes basal ganglia and claustrum
- CSF: often normal or mild pleocytosis
- May mimic Creutzfeldt-Jakob disease (rapid cognitive decline + myoclonic movements)
Mechanism:
Treatment Response:
- ~80% have substantial response to immunotherapy
- Relapses in 27-35%
3. CASPR2 (Contactin-Associated Protein-Like 2) - VGKC Complex Glycoprotein
What is CASPR2?
Who gets it?
- Elderly men predominantly (similar to LGI1 but slightly older)
- ~20-50% have underlying thymoma (higher malignancy association than LGI1)
- IgG4 isotype (unique among autoimmune encephalitides)
Clinical Features - Both CNS and PNS Involved:
| Syndrome | Features |
|---|---|
| Limbic encephalitis | Memory loss, confusion, temporal lobe seizures |
| Cerebellar dysfunction | Ataxia |
| Peripheral nerve hyperexcitability (PNH) | Myokymia, cramps, fasciculations, hyperhidrosis |
| Morvan Syndrome | CNS symptoms + PNH + autonomic dysfunction + severe insomnia (agrypnia excitata) |
| Neuropathic pain | Allodynia, painful sensory disturbances |
Mechanism:
Important Association:
4. GLYCINE RECEPTOR (GlyR) Antibodies
- Target: Glycine receptor - a ligand-gated chloride channel glycoprotein
- Syndrome: PERM (Progressive Encephalomyelitis with Rigidity and Myoclonus) + limbic encephalitis
- Features: muscle stiffness, hyperekplexia (exaggerated startle), brainstem dysfunction
- Rare association with thymoma
Complete Classification: Glycoprotein/Surface Antigen Targets in Autoimmune Encephalitis
| Antibody Target | Type | Demographics | Hallmark Feature | Cancer Association |
|---|---|---|---|---|
| NMDA receptor | Ion channel | Young females | Psychiatric prodrome → seizures → autonomic instability → coma | Ovarian teratoma (50%) |
| LGI1 | Secreted glycoprotein (VGKC complex) | Elderly males | Faciobrachial dystonic seizures + hyponatremia | <10%, thymoma |
| CASPR2 | Transmembrane glycoprotein (VGKC complex) | Elderly males | Morvan syndrome, PNH + CNS | Thymoma (20-50%) |
| MOG | Myelin glycoprotein | Young, children | Bilateral ON, ADEM, cortical encephalitis | Rare |
| AMPA receptor | Ion channel | Middle-aged women | Limbic encephalitis, relapses | Lung, breast, thymus (60%) |
| GABA-B receptor | Metabotropic receptor | Elderly | Refractory seizures, ataxia | Small cell lung (50%) |
| GABA-A receptor | Ion channel | Any age | Refractory SE, multifocal MRI lesions | Thymoma (infrequent) |
| Glycine receptor | Ligand-gated channel | Any age | PERM, hyperekplexia | Thymoma (rare) |
| DPPX | Glycoprotein | Middle age | Diarrhea/weight loss prodrome + hyperekplexia | Rare B-cell neoplasm |
Treatment Approach for Glycoprotein-Positive Autoimmune Encephalitis
- First-line (acute): IV methylprednisolone + IVIG + plasmapheresis (in combination)
- If no response (4-6 weeks): Rituximab or cyclophosphamide
- Maintenance: Azathioprine, mycophenolate mofetil, or rituximab infusions
- Tumor treatment: If paraneoplastic - tumor removal is itself therapeutic and improves neurological outcome
Treatment for the congentinal myasthenia gravis
congenital myasthenic syndrome treatment pyridostigmine salbutamol ephedrine 3,4-DAP subtype specific 2024
Treatment of Congenital Myasthenic Syndromes (CMS)
Important Distinction First
| Feature | Autoimmune MG | Congenital Myasthenic Syndrome |
|---|---|---|
| Cause | Autoantibodies (AChR, MuSK, LRP4) | Gene mutations |
| Onset | Any age | Birth or childhood (usually) |
| Antibody testing | Positive in 85-90% | Negative |
| Immunosuppression | Central treatment | Ineffective and should be AVOIDED |
| Treatment | Pyridostigmine + immunosuppression + thymectomy | Genotype-specific pharmacotherapy |
Differentiating CMS from seronegative autoimmune MG is critical so that ineffective immunosuppressive therapies can be avoided. Genetic analysis is the key diagnostic test. (Goldman-Cecil Medicine)
The Golden Rule: Treatment is SUBTYPE-SPECIFIC
- Cholinesterase inhibitors (pyridostigmine) are HARMFUL in slow-channel CMS and AChE deficiency
- Quinidine and fluoxetine are HARMFUL in fast-channel CMS
- DOK-7 CMS worsens with pyridostigmine
Treatment by CMS Subtype
A. PRESYNAPTIC SUBTYPES (Deficient ACh release)
1. ChAT Deficiency (Choline Acetyltransferase deficiency) - "Episodic Apnea"
- Defect: ChAT enzyme absent or reduced → impaired ACh synthesis
- Clinical: Neonatal hypotonia, episodic apneic crises (precipitated by fever/stress), mild baseline weakness
- Treatment:
- AChE inhibitors (pyridostigmine) - first-line; prolongs ACh lifetime at synapse
- Apnea monitor mandatory (risk of sudden death)
- Symptoms tend to lessen in adolescence
2. Reduced Quantal ACh Release (Unknown cause)
- Treatment: AChE inhibitors + 3,4-Diaminopyridine (3,4-DAP / amifampridine)
B. SYNAPTIC SUBTYPES (Endplate enzyme defects)
3. AChE Deficiency (COLQ mutation)
- Defect: No collagenous tail (COLQ) for anchoring AChE at endplate → ACh not degraded → prolonged depolarization → endplate damage
- Clinical: Severe generalized weakness, ptosis, ophthalmoparesis, slowed pupillary responses, repetitive discharges on EMG after single nerve stimulus
- Treatment:
- Ephedrine or Salbutamol (β2-adrenergic agonists) - first-line
- AVOID AChE inhibitors (pyridostigmine worsens this condition - ACh is already not being degraded, adding more ACh to the already excessive stimulation worsens endplate damage)
4. DOK-7 Synopathy (DOK-7 mutation)
- Defect: DOK-7 activates MuSK → failure of AChR clustering and endplate development
- Clinical: Limb-girdle pattern of weakness (may be mistaken for muscular dystrophy), reduced fetal movements in utero
- Treatment:
- Ephedrine, Salbutamol, or Albuterol - first-line β2-agonists
- Amifampridine may provide additional benefit
- AVOID AChE inhibitors - worsen the condition
C. POSTSYNAPTIC SUBTYPES (AChR kinetic defects)
5. Slow-Channel Syndrome (SCCMS)
- Defect: AChR subunit mutations (CHRNE, CHRNA, CHRNB, CHRND genes) → channel stays open too long → excessive Ca²⁺ entry → endplate myopathy and degeneration
- Inheritance: Autosomal dominant (only CMS with dominant inheritance)
- Clinical: Neck and distal upper limb weakness (hand/finger extensors), ptosis; onset ranges from infancy to 7th decade; EMG shows repetitive muscle discharges after single stimulus
- Treatment:
- Quinidine sulfate - blocks the prolonged channel opening; improves strength
- Fluoxetine - also reduces channel open time (open-channel blocker)
- AVOID AChE inhibitors - prolong ACh action → more channel opening → worsens endplate degeneration
6. Fast-Channel Syndrome
- Defect: AChR subunit mutations → channel opens too briefly → insufficient depolarization → severely reduced safety margin
- Clinical: Usually present at birth, severe ptosis, ophthalmoplegia, weak cry, poor feeding, respiratory crises; can be fatal without support
- Treatment:
- Ventilator support (often from birth) - primary supportive care
- Gastrostomy for feeding
- AChE inhibitors (pyridostigmine) + Amifampridine (3,4-DAP) - both help prolong/augment ACh availability
- Improvement from AChE inhibitors may wane over time
7. Primary AChR Deficiency (ε-subunit mutations, CHRNE, CHRNA, etc.)
- Defect: Reduced AChR expression at endplate → reduced safety margin for transmission
- Clinical: Ptosis, limb weakness, fatigability; most common CMS subtype in UK
- Treatment:
- AChE inhibitors (pyridostigmine) - first-line
- 3,4-DAP (amifampridine) - prolongs presynaptic depolarization → more ACh released per impulse
- Salbutamol or ephedrine - additional benefit; β2-agonists upregulate AChR expression
8. Rapsyn Deficiency (RAPSN mutations)
- Defect: Rapsyn clusters AChRs at endplate; mutations → sparse, poorly organized AChRs
- Clinical: Respiratory distress at birth, hypotonia, generalized weakness, ptosis (ophthalmoplegia uncommon), arthrogryposis, high-arched palate; respiratory crises until ~age 7
- Treatment:
- AChE inhibitors + Amifampridine (3,4-DAP) - good response
- Salbutamol may add benefit
- Prognosis is relatively favorable - many patients can discontinue treatment as adults
9. GFPT1/DPAGT1 Mutations (Glycosylation defects)
- Defect: Impaired glycosylation of NMJ proteins → limb-girdle weakness
- Clinical: Progressive limb-girdle weakness beginning in childhood/early teens; minimal ocular/bulbar involvement; EMG shows myopathic + NMJ abnormalities
- Treatment:
- AChE inhibitors + Amifampridine - benefit most patients
- Albuterol/ephedrine may also help
Drug Mechanisms in CMS
| Drug | Class | Mechanism | Used for |
|---|---|---|---|
| Pyridostigmine | AChE inhibitor | Inhibits acetylcholinesterase → more ACh at synapse, longer action | Presynaptic CMS, AChR deficiency, rapsyn, fast-channel |
| 3,4-DAP (amifampridine) | K⁺ channel blocker | Blocks presynaptic K⁺ channels → prolonged depolarization → more Ca²⁺ entry → more ACh vesicle release | Presynaptic CMS, AChR deficiency, rapsyn, fast-channel |
| Salbutamol / Albuterol | β2-agonist | Upregulates AChR expression; stabilizes NMJ morphology | DOK-7, AChE deficiency, AChR deficiency, COLQ |
| Ephedrine | Sympathomimetic (α+β) | Similar to salbutamol; also increases ACh release | DOK-7, AChE deficiency |
| Quinidine | Open-channel blocker | Reduces prolonged AChR channel open time | Slow-channel CMS only |
| Fluoxetine | SSRI / Open-channel blocker | Reduces AChR channel open time (same mechanism as quinidine) | Slow-channel CMS only |
Transient Neonatal MG - Important Differential
Summary Treatment Table
| Subtype | Location | Treatment | AVOID |
|---|---|---|---|
| ChAT deficiency | Presynaptic | Pyridostigmine + apnea monitor | - |
| Reduced quantal release | Presynaptic | Pyridostigmine + 3,4-DAP | - |
| AChE deficiency (COLQ) | Synaptic | Ephedrine / Salbutamol | Pyridostigmine |
| DOK-7 | Synaptic | Ephedrine / Salbutamol | Pyridostigmine |
| Slow-channel | Postsynaptic | Quinidine / Fluoxetine | Pyridostigmine |
| Fast-channel | Postsynaptic | Ventilator + Pyridostigmine + 3,4-DAP | - |
| AChR deficiency | Postsynaptic | Pyridostigmine + 3,4-DAP + Salbutamol | - |
| Rapsyn deficiency | Postsynaptic | Pyridostigmine + 3,4-DAP | - |
| GFPT1/DPAGT1 | Postsynaptic | Pyridostigmine + 3,4-DAP + Salbutamol | - |
Why the vanco and ceftrixone combination are used in the meningities What is the tx for the vanco and ceftrixone resistance
vancomycin ceftriaxone resistant meningitis treatment meropenem rifampicin linezolid 2024 guidelines
Part 1: Why Vancomycin + Ceftriaxone in Meningitis
- Ceftriaxone covers susceptible S. pneumoniae, N. meningitidis, H. influenzae, Group B streptococci - excellent CSF penetration through inflamed meninges
- Vancomycin covers penicillin/cephalosporin-resistant S. pneumoniae - the organism most likely to cause treatment failure and death
- Neither drug alone gives complete coverage: ceftriaxone fails against resistant pneumococcus; vancomycin alone has poor/variable CSF penetration and no Gram-negative coverage
- Together they are synergistic and cover the full spectrum of common community-acquired meningitis organisms before culture results are available
Part 2: Treatment When Vancomycin + Ceftriaxone Resistance Occurs
Step 1: Confirm Resistance - Repeat LP at 24-36 Hours
Resistance Scenarios and Treatment
SCENARIO A: Pneumococcal Meningitis - Cephalosporin-Resistant but Vancomycin-Susceptible
| Pneumococcal MIC | Interpretation | Treatment |
|---|---|---|
| Penicillin MIC <0.06 μg/mL | Susceptible | Penicillin G (narrow down) |
| Penicillin MIC 0.06-0.12 μg/mL | Intermediate | 3rd-generation cephalosporin |
| Cefotaxime/ceftriaxone MIC ≤0.5 μg/mL | Susceptible | Ceftriaxone alone adequate |
| Cefotaxime/ceftriaxone MIC = 1 μg/mL | Intermediate | Vancomycin = drug of choice |
| Cefotaxime/ceftriaxone MIC ≥ 2 μg/mL | Resistant | Vancomycin + Ceftriaxone (continue) ± Rifampicin |
SCENARIO B: Pneumococcal Meningitis Failing Vancomycin + Ceftriaxone
1. Add Rifampicin (Rifampin) - First Escalation Step
- Mechanism: Inhibits bacterial RNA polymerase; excellent CSF penetration; acts synergistically with vancomycin and cephalosporins against pneumococcus
- Dose: 600 mg IV/PO q12-24h (adults); 10 mg/kg q12h (children)
- Critical rule: NEVER use rifampicin as monotherapy - resistance develops rapidly when used alone
- Indication: Add when clinical or bacteriologic response is delayed; also useful when dexamethasone reduces vancomycin CSF penetration
- As Harrison's states: "Rifampin can be added to vancomycin for its synergistic effect but is inadequate as monotherapy because resistance develops rapidly when it is used alone"
2. Intraventricular or Intrathecal Vancomycin
- When IV vancomycin fails to achieve adequate CSF levels (especially with concurrent dexamethasone)
- Intraventricular route preferred over intrathecal - more reliable CSF distribution; adequate concentrations in cerebral ventricles not always achieved intrathecally
- Dose: 20 mg intraventricular once daily (via external ventricular drain)
- Requires neurosurgical consultation
3. Meropenem - Alternative β-Lactam
- Mechanism: Carbapenem; inhibits PBPs with activity against penicillin-resistant pneumococcus
- CSF penetration: Good through inflamed meninges
- Role: Alternative to ceftriaxone in β-lactam allergic patients; also used for Gram-negative meningitis including Pseudomonas
- Limitation: In experimental models, meropenem was comparable to ceftriaxone but inferior to vancomycin against resistant pneumococcus
- Indication: When cephalosporins are contraindicated (allergy) OR for nosocomial meningitis with Gram-negatives
4. Linezolid - Reserved for Multi-Drug Resistance
- Mechanism: Oxazolidinone; inhibits 50S ribosomal protein synthesis; bacteriostatic against pneumococcus
- CSF penetration: Good (80% of serum levels)
- Dose: 600 mg IV/PO q12h
- Use: Cephalosporin-resistant pneumococcal meningitis; MRSA meningitis; VRE meningitis
- Rosen's EM explicitly states: "Linezolid (600 mg every 12 hours) with vancomycin can be used in cephalosporin-resistant strains of pneumococcus"
- Limitation: Bacteriostatic (not bactericidal); long-term use causes thrombocytopenia, peripheral neuropathy, serotonin syndrome
5. Moxifloxacin - Fluoroquinolone Option
- Mechanism: Inhibits DNA gyrase and topoisomerase IV; excellent CNS penetration
- Dose: 400 mg IV/PO once daily
- Use: Cephalosporin-resistant pneumococcal meningitis; penicillin-allergic patients
- Rosen's EM: "Moxifloxacin (400 mg once daily) with vancomycin can be used in cephalosporin-resistant strains"
- Caution: QT prolongation; avoid in patients already on QT-prolonging drugs
6. Chloramphenicol - Historic Alternative
- Good CSF penetration even through non-inflamed meninges
- Bactericidal against pneumococcus, meningococcus, H. influenzae
- Now rarely used due to toxicity (aplastic anemia, grey baby syndrome) and increasing resistance
- Still used in low-income countries and as a β-lactam/vancomycin allergy backup
SCENARIO C: MRSA Meningitis (Post-Neurosurgery / Nosocomial)
| First-line | Alternative |
|---|---|
| Vancomycin (high dose, target AUC/MIC) | Linezolid, daptomycin, TMP-SMX |
| + Rifampicin (if susceptible) | |
| If CSF not sterile in 48h → add intraventricular/intrathecal vancomycin 20 mg OD |
SCENARIO D: Gram-Negative Bacillary Meningitis Resistance (e.g., ESBL/Carbapenem)
| Organism | Preferred | Alternative |
|---|---|---|
| Susceptible Enterobacteriaceae | Ceftriaxone/cefotaxime | Meropenem, aztreonam, TMP-SMX |
| ESBL-producing Enterobacteriaceae | Meropenem | Aztreonam |
| Pseudomonas aeruginosa | Ceftazidime or cefepime | Meropenem, aztreonam, ciprofloxacin |
| Carbapenem-resistant Acinetobacter | Meropenem + colistin/polymyxin B | |
| Carbapenem-resistant Pseudomonas | Colistin/polymyxin B ± intrathecal colistin |
Decision Algorithm for Failed Vanco + Ceftriaxone
Vanco + Ceftriaxone started → Repeat LP at 24-36h
↓
CSF still culture-positive?
↓
┌─────────────────────────────────────────┐
│ 1. Add RIFAMPICIN (synergy, no monotherapy) │
│ 2. Intraventricular VANCOMYCIN if low CSF vanco │
│ 3. Switch to MEROPENEM if β-lactam allergy │
│ 4. LINEZOLID + vancomycin (resistant cocci) │
│ 5. MOXIFLOXACIN (fluoroquinolone option) │
└─────────────────────────────────────────┘
↓
Infectious Disease Consultation MANDATORY
+ Neurosurgery for intraventricular access
Summary Table: Alternatives to Vanco + Ceftriaxone
| Scenario | Drug | Key Point |
|---|---|---|
| Ceph-resistant S. pneumoniae | + Rifampicin | Never monotherapy |
| Failed IV vancomycin | Intraventricular vancomycin 20 mg OD | Preferred over intrathecal |
| β-Lactam allergy | Meropenem | Good Gram+ and Gram- cover |
| Multi-resistant pneumococcus | Linezolid 600 mg q12h | Bacteriostatic; watch toxicity |
| Alternative to cefts | Moxifloxacin 400 mg OD | QT prolongation risk |
| MRSA meningitis | Vancomycin ± Rifampicin ± intrathecal | High-dose IV |
| Pseudomonas meningitis | Ceftazidime or Meropenem | Not ceftriaxone |
| Resistant Gram-negative | Meropenem ± colistin | Carbapenem backbone |
How do we takle the vancomycin plus the dexamethasone
vancomycin dexamethasone interaction meningitis CSF penetration management higher dose intraventricular 2024
Vancomycin + Dexamethasone in Meningitis - The Core Problem and Its Solution
The Problem: Dexamethasone Tightens the Blood-Brain Barrier
| Effect | Dexamethasone Action | Consequence |
|---|---|---|
| Benefit | Inhibits IL-1β and TNF-α at mRNA level → reduces neuroinflammation | Less brain edema, less sensorineural hearing loss, lower mortality |
| Benefit | Decreases CSF outflow resistance | Lowers ICP |
| Benefit | Stabilizes the blood-brain barrier (BBB) | Reduces vascular leak |
| Problem | Same BBB stabilization that is beneficial also reduces permeability to vancomycin | Vancomycin CSF penetration drops → risk of undertreating resistant S. pneumoniae |
What Does the Clinical Evidence Actually Show?
- Enrolled patients with suspected S. pneumoniae meningitis treated with both empiric antibiotics and dexamethasone
-
50% had confirmed penicillin-resistant S. pneumoniae
- Result: All patients had CSF vancomycin concentrations at least 4-fold above the MIC of their cultured organism
- On repeat LP: none had positive S. pneumoniae cultures
- CSF vancomycin levels were proportional to serum vancomycin levels (i.e., serum monitoring still predicts CSF levels)
The Solution: Three Strategies
Strategy 1: Increase the Vancomycin Dose - The Primary Solution
"Dexamethasone may decrease the penetration of vancomycin into CSF... as a result, to assure reliable penetration of vancomycin into the CSF, children and adults are treated with vancomycin in a dose of 45-60 mg/kg per day." - Harrison's Principles of Internal Medicine, 22nd ed.
| Patient | Standard Dose | With Dexamethasone |
|---|---|---|
| Adults | 30 mg/kg/day (15 mg/kg q12h) | 45-60 mg/kg/day (divided q8-12h) |
| Children | 40-60 mg/kg/day | 60 mg/kg/day (q6h) |
Strategy 2: Monitor Serum Vancomycin Levels - AUC/MIC Targeting
- Target: AUC/MIC ratio of 400-600 (current AUC-guided monitoring, replacing old trough-only monitoring)
- Serum trough target (if AUC monitoring not available): 15-20 mg/L
- Continuous infusion achieves more stable CSF levels than intermittent bolus - RCT data shows higher mean CSF concentrations with continuous infusion of 50 mg/kg/day vs. intermittent bolus
- TDM (therapeutic drug monitoring) is mandatory when dexamethasone is co-administered
Strategy 3: Intraventricular Vancomycin - When IV Fails
- Intraventricular vancomycin: 20 mg once daily via external ventricular drain (EVD)
- Preferred over intrathecal (lumbar intrathecal) because:
- Intrathecal injection in lumbar space does not reliably distribute to cerebral ventricles
- Drug may pool in lumbar CSF without reaching ventricular compartment where bacteria may reside
- Intraventricular = direct drug delivery into ventricular system → reliable distribution
- Requires neurosurgical consultation for EVD placement
Do You Still Give Dexamethasone?
| Benefit | Data |
|---|---|
| Reduced unfavorable outcomes in pneumococcal meningitis | 15% vs 25% (p=0.03), European RCT |
| Reduced death in pneumococcal meningitis | 7% vs 15% (p=0.04) |
| Reduced sensorineural hearing loss | Consistent across trials |
| Reduced hearing loss in H. influenzae meningitis | Established, especially in children |
- Listeria meningitis identified - dexamethasone associated with increased adverse outcomes (large prospective study)
- Sub-Saharan Africa / low-income countries with negative CSF Gram stain and culture - no benefit shown in three large RCTs (attributed to late presentation, HIV, malnutrition, antibiotic pretreatment)
- HIV-associated cryptococcal meningitis - dexamethasone causes harm (NEJM 2016)
Practical Clinical Protocol Summary
Suspected bacterial meningitis
↓
1. Draw blood cultures (immediately)
2. Dexamethasone 10 mg IV (15-20 min before antibiotics)
↓
3. Vancomycin 45-60 mg/kg/day IV (q8-12h) - HIGH DOSE because of dexa
+ Ceftriaxone 2g q12h
(+ Ampicillin 2g q4h if age >55 or immunocompromised → Listeria)
↓
4. Continue dexamethasone 10 mg q6h × 4 days
→ STOP if Listeria confirmed
↓
5. Repeat LP at 24-36 hours → confirm CSF sterilization
↓
6. Monitor vancomycin serum AUC/MIC (target 400-600)
↓
7. If CSF not sterile at 24-36h:
→ Add rifampicin 600 mg q12h
→ Consider intraventricular vancomycin 20 mg OD
→ ID consultation
Key Points Summary
| Question | Answer |
|---|---|
| Does dexamethasone reduce vancomycin CSF levels? | Yes, by ~29% in animal models |
| Is this clinically significant at standard doses? | Potentially yes - concern supported by case reports |
| How do you compensate? | Increase vancomycin to 45-60 mg/kg/day |
| How do you monitor? | AUC/MIC targeting; serum trough 15-20 mg/L |
| What if IV vancomycin still fails? | Intraventricular vancomycin 20 mg OD via EVD |
| Should you still give dexamethasone? | Yes - mortality/morbidity benefit > PK concern |
| When to stop dexamethasone? | If Listeria confirmed; if low-income setting with no culture |
| Best timing for dexamethasone? | 15-20 min before first antibiotic dose |
Dosing strategies for antibiotics with dexamethasone in pediatric meningitis
pediatric bacterial meningitis antibiotic dosing dexamethasone vancomycin 60mg/kg children 2024 guidelines
https://cps.ca/en/documents/position/management-of-bacterial…
Antibiotic Dosing Strategies for Pediatric Meningitis + Dexamethasone
Part 1: Age-Stratified Empiric Antibiotic Selection
Neonates <1 Month (0-28 days)
| Drug | Dose | Interval | Notes |
|---|---|---|---|
| Ampicillin | 300 mg/kg/day | q6h (or q8h if <7 days old: 225 mg/kg/day) | Covers GBS + Listeria - essential |
| Cefotaxime | 150-200 mg/kg/day | q6-8h | Covers Gram-negatives (E. coli, Klebsiella) |
| OR Gentamicin | 5 mg/kg/day | q24h (once daily) | Added if Gram-negative rods on Gram stain |
| Acyclovir | 60 mg/kg/day | q8h (20 mg/kg/dose) | Add empirically if HSV risk: vesicles, seizures, CSF pleocytosis with negative Gram stain |
- NEVER use ceftriaxone in neonates - especially in hyperbilirubinemia - ceftriaxone displaces bilirubin from albumin binding sites → worsens jaundice and risks kernicterus. Use cefotaxime instead
- Vancomycin in neonates: Add only in late-onset nosocomial disease or infected VP shunt: 45 mg/kg/day divided q8h (30 mg/kg/day for age <7 days)
- Cephalosporins do NOT cover Listeria - ampicillin is mandatory
- Dexamethasone is NOT recommended in neonates - no benefit shown; risk of intestinal perforation; immature HPA axis
Infants 1-3 Months (Transitional Period)
| Drug | Dose | Interval |
|---|---|---|
| Ampicillin | 300 mg/kg/day | q6h |
| Cefotaxime | 200-300 mg/kg/day | q6h |
| Vancomycin | 60 mg/kg/day | q6h |
Infants/Children >3 Months to 18 Years - Standard Pediatric Regimen
| Drug | Dose | Interval | Max Daily Dose |
|---|---|---|---|
| Vancomycin | 60 mg/kg/day | q6h | 4 g/day (2 g/dose) |
| Ceftriaxone | 100 mg/kg/day | q12h | 4 g/day |
| OR Cefotaxime | 200-300 mg/kg/day | q6h | 8-12 g/day |
"For infants and children with meningitis, vancomycin 60 mg/kg/day in divided doses every 6h to achieve trough concentrations of 10-15 mg/L." - Canadian Paediatric Society Guidelines
Part 2: The Vancomycin + Dexamethasone Dosing Problem in Children
Why Children Need Higher Vancomycin Doses Than Adults
- Higher renal clearance per kg in children → faster drug elimination
- Larger volume of distribution relative to body weight
- Faster metabolism
- Adult meningitis dose WITHOUT dexamethasone: 30 mg/kg/day
- Adult meningitis dose WITH dexamethasone: 45-60 mg/kg/day
- Pediatric meningitis dose (always): 60 mg/kg/day q6h - this dose was specifically validated to achieve therapeutic CSF levels even when dexamethasone is given concurrently
Part 3: Dexamethasone Dosing and Indications in Children
Dose
| Source | Dose | Schedule | Duration |
|---|---|---|---|
| CPS Guidelines | 0.6 mg/kg/day divided q6h = 0.15 mg/kg/dose q6h | q6h × 4 days | 4 days |
| Harrison's / Rosen's | 0.15 mg/kg/dose q6h (max 10 mg/dose) | q6h × 4 days | 4 days |
Timing - The Non-Negotiable Rule
- Give 15-20 minutes BEFORE or at the same time as the first antibiotic dose
- Within 4 hours of first antibiotic - benefit likely decreasing
- After 6 hours of antibiotics - NO benefit (TNF-α and IL-1β already released and cannot be suppressed)
- Do NOT delay antibiotics to obtain dexamethasone - if dexamethasone is not immediately available, give antibiotics first
When to Give Dexamethasone in Children
| Situation | Recommendation |
|---|---|
| H. influenzae type b (Hib) meningitis | Strongly recommended - best evidence; reduces hearing loss and neurologic sequelae |
| S. pneumoniae meningitis | Recommended (most guidelines); reduces hearing loss and mortality |
| N. meningitidis | Consider - some benefit for hearing loss; mortality benefit unproven |
| Gram-negative bacillary meningitis (neonates/infants) | Not recommended |
| Neonates <6 weeks | Not recommended - no benefit; risk of GI perforation |
| Unknown organism, strong clinical suspicion | Give empirically → reassess at 48h |
When to STOP Dexamethasone at 48 Hours
| Finding at 48h | Action |
|---|---|
| Hib confirmed | Continue full 4 days |
| Pneumococcus confirmed | Continue full 4 days |
| Hib NOT identified on culture/PCR | STOP dexamethasone |
| Listeria confirmed | STOP immediately - dexamethasone associated with worse outcomes |
| N. meningitidis only | Discontinuation reasonable; benefit not proven |
| Viral meningitis (negative culture) | STOP |
Part 4: Full Pediatric Antibiotic Dosing Reference Table
| Drug | Pediatric Dose | Max Daily Dose | Interval | Notes |
|---|---|---|---|---|
| Vancomycin | 60 mg/kg/day (60-70 with dexa) | 4 g/day | q6h | Trough target 10-15 mg/L; AUC/MIC 400-600 |
| Ceftriaxone | 100 mg/kg/day | 4 g/day | q12h | Avoid neonates (bilirubin displacement) |
| Cefotaxime | 200-300 mg/kg/day | 8-12 g/day | q6h | Safe in neonates; preferred <1 month |
| Ampicillin | 300-400 mg/kg/day | 6-12 g/day | q6h | Listeria + GBS coverage; mandatory <3 months |
| Penicillin G | 300,000-400,000 units/kg/day | 24 million units/day | q4-6h | Once pathogen confirmed susceptible |
| Gentamicin | 5 mg/kg/day | - | q24h | Synergy with ampicillin for GBS/Listeria in critically ill |
| Meropenem | 120 mg/kg/day | 4-6 g/day | q8h | β-lactam allergy; resistant Gram-negatives; Pseudomonas |
| Ceftazidime | 150 mg/kg/day | 6 g/day | q8h | Pseudomonas aeruginosa meningitis |
| Cefepime | 150 mg/kg/day | 2-4 g/day | q8h | Limited pediatric data; not licensed for meningitis |
| Rifampicin | 20 mg/kg/day | 600 mg/day | q12h | Add to vancomycin if response delayed; never monotherapy |
| Acyclovir | 60 mg/kg/day | - | q8h (20 mg/kg/dose) | HSV encephalitis/meningitis in neonates |
| Dexamethasone | 0.6 mg/kg/day | 40 mg/day | q6h × 4 days | 15-20 min BEFORE first antibiotic |
Part 5: Therapeutic Drug Monitoring (TDM) for Vancomycin in Children with Dexamethasone
| Parameter | Target | Notes |
|---|---|---|
| Trough level (traditional) | 10-15 mg/L for meningitis | Check 30 min before 4th dose |
| AUC/MIC (modern, preferred) | 400-600 | Requires Bayesian software or 2 serum levels |
| Peak level | Not routinely targeted | Less useful for meningitis TDM |
| Maximum single dose | 2 g/dose | Do not exceed regardless of weight |
| Maximum daily dose | 4 g/day | Total cap |
| Nephrotoxicity monitoring | SCr at baseline and q48-72h | Particularly important with aminoglycosides co-admin |
Summary Protocol for Child >3 Months with Suspected Bacterial Meningitis
Suspected bacterial meningitis in child >3 months
↓
Blood cultures (immediate) → LP if safe (no papilledema/focal deficits)
↓
Give simultaneously:
1. Dexamethasone 0.15 mg/kg IV (max 10 mg) → 15-20 min before antibiotics
2. Vancomycin 60-70 mg/kg/day IV (q6h, max 2g/dose) ← HIGH dose because of dexa
3. Ceftriaxone 100 mg/kg/day IV (q12h, max 4g/day)
[If <3 months or immunocompromised → add Ampicillin 300mg/kg/day q6h]
↓
Continue dexamethasone 0.15 mg/kg q6h × 4 days TOTAL
↓
Check vancomycin trough before 4th dose → target 10-15 mg/L
↓
Repeat LP at 24-36h → confirm CSF sterilization
↓
At 48h: culture results available
→ Hib confirmed: continue 4 days dexa, narrow antibiotics
→ Pneumococcus: continue 4 days dexa, check susceptibility, narrow
→ Listeria: STOP dexamethasone, treat with ampicillin
→ Meningococcus: consider stopping dexa (benefit unproven)
→ No organism identified: stop dexa
↓
If CSF not sterile at 24-36h → Add rifampicin 20 mg/kg/day q12h
If IV vancomycin failing → Intraventricular vancomycin
(5 mg for slit ventricles / 10 mg normal / 15-20 mg enlarged)
What is the treatment for after administrative of antibiotics after 6 hrs with out the dexamethasone and how can we manage them
dexamethasone missed delayed after 6 hours antibiotics meningitis management supportive care complications 2024
What are the complications of missed dexamethasone in meningitis and their management?