Glucaconoma
Reading File
Finding Sources
Searching PubMed
glucagonoma syndrome pancreatic neuroendocrine tumor
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Here is a thorough summary:
Glucagonoma
Glucagonoma is a rare functioning pancreatic neuroendocrine tumor (PNET) arising from the alpha cells of the islets of Langerhans. It secretes excess glucagon and produces a distinctive clinical syndrome. Its incidence is approximately 1 in 10 million population.
Pathophysiology
Glucagon is a catabolic hormone that, in excess, drives:
- Hepatic glycogenolysis and gluconeogenesis (causing hyperglycemia)
- Ketogenesis and lipolysis
- Inhibition of pancreatic and gastric secretions
- Inhibition of gastrointestinal motility
These effects collectively lead to severe catabolism, malnutrition, and protein wasting. Hypoaminoacidemia (depletion of plasma amino acids) is thought to be a key trigger for the characteristic rash.
Clinical Features ("4 Ds" + more)
| Feature | Details |
|---|---|
| Dermatitis (NME) | Present in ~70% at diagnosis; eventually near-universal |
| Diabetes mellitus | Glucose intolerance in 22-90%; usually precedes diagnosis by ~5 years |
| DVT / Thromboembolism | Risk 4-33%; higher than any other NET |
| Depression / psychiatric | Reported psychiatric disturbances |
| Weight loss | Profound weight loss in 54-83% |
| Anemia | Normocytic, normochromic |
| Hypoaminoacidemia | Hallmark biochemical finding |
| Hypocholesterolemia | Also characteristic |
| Glossitis, stomatitis, cheilitis | Mucosal involvement |
Necrolytic Migratory Erythema (NME) - The Classic Rash
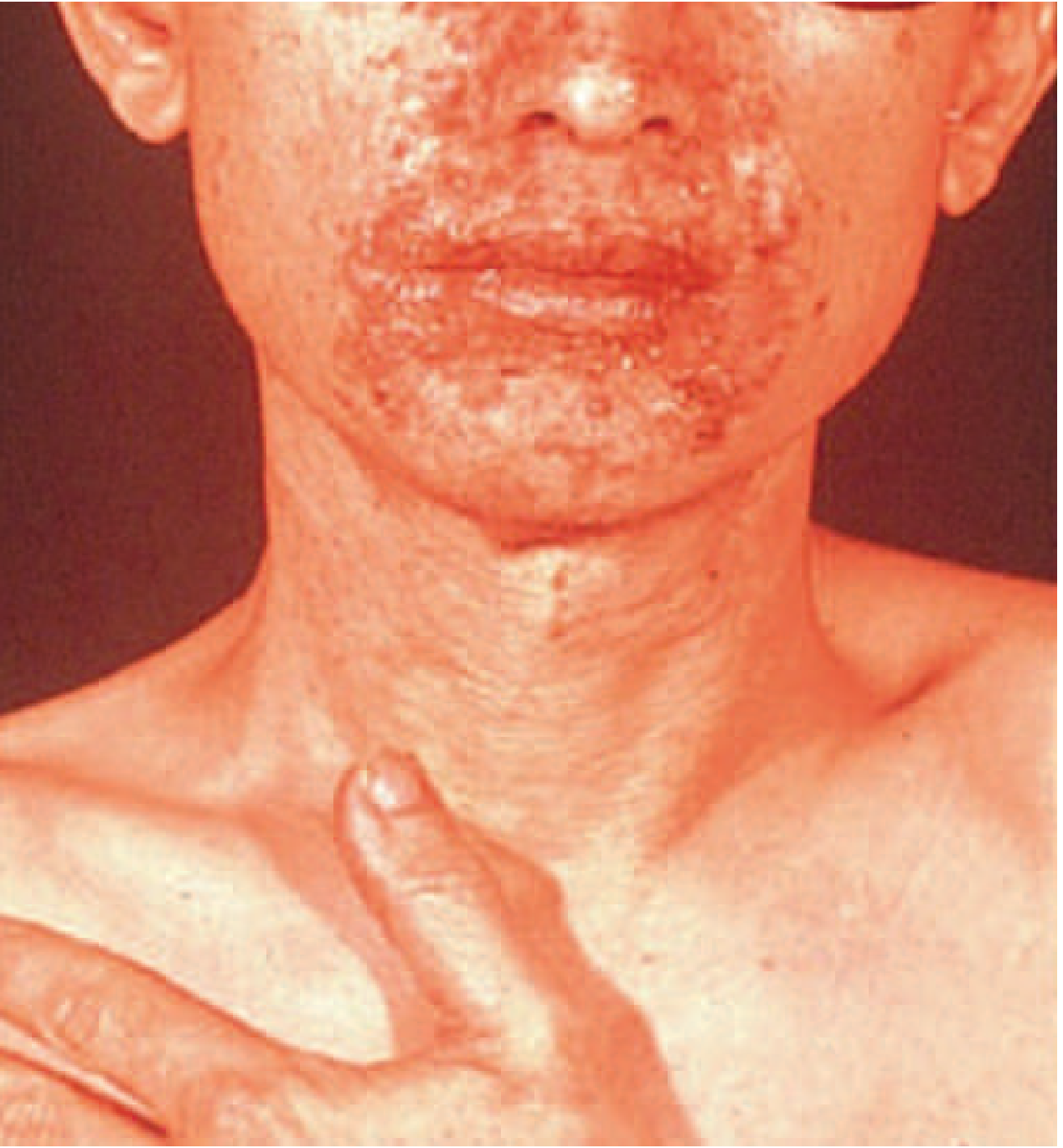
Classic NME involving the perioral and perifacial region in a patient with glucagonoma syndrome.
The rash follows a characteristic cycle of 1-2 weeks:
- Erythematous macules and papules at perioral/intertriginous sites (groin, buttocks, perineum)
- Expansion into confluent plaques with superficial scale
- Central superficial bullae that rupture, forming erosions
- Crusting and healing with hyperpigmentation
- Lesions appear asynchronously - areas at various stages simultaneously
- Relapsing and remitting course
The rash often precedes the diagnosis of glucagonoma by years and is frequently misdiagnosed. NME is not pathognomonic - it also occurs in myelodysplastic syndrome, short gut syndrome, hepatitis B, cirrhosis, celiac disease, and severe malnutrition.
Epidemiology & Tumor Characteristics
- Age of onset: primarily >50 years, slight female predominance
- ~90% sporadic; ~10% associated with MEN1
- Almost exclusively pancreatic in origin (vs other NETs)
- Located predominantly in the tail of the pancreas
- Single tumor in the vast majority
- Large at diagnosis: >60% are >3 cm in diameter
- Metastatic at presentation in 42-78% (most commonly to liver, lymph nodes, bone, mesentery)
Diagnosis
Gold standard: Fasting plasma glucagon level
| Level | Interpretation |
|---|---|
| Normal | <120 pg/mL |
| Mild elevation | Associated with stress, cirrhosis, DKA, renal failure, acromegaly, burns, etc. |
| Glucagonoma | Typically >500 pg/mL (Harrison's, Yamada's); >1000 pg/mL (Harrison's ch. 89) |
Supporting findings:
- Hypoaminoacidemia
- Normocytic anemia
- Hypocholesterolemia
- Hyperglycemia / diabetic pattern
Imaging: CT/MRI to localize the pancreatic mass and assess for metastases. Somatostatin receptor scintigraphy (e.g., Ga-68 DOTATATE PET) is also used for functional imaging.
Treatment
Surgical
- Distal pancreatectomy + splenectomy for localized tail lesions (curative intent)
- Resection even with metastatic disease may be considered (debulking)
- Pre-op: Amino acid infusion or total parenteral nutrition (TPN) improves the dermatologic symptoms and nutritional status before surgery
- Perioperative anticoagulation is recommended given the markedly elevated VTE risk (DVT/PE in up to 33%)
Medical (especially for metastatic/inoperable disease)
- Somatostatin analogues (SSAs) - octreotide or lanreotide: usually highly effective in controlling the rash and hormonal symptoms
- Chemotherapy: streptozotocin + 5-fluorouracil for metastatic disease
- Hepatic artery embolization for liver metastases
- Everolimus or sunitinib (approved for advanced PNETs)
Nutritional
- Amino acid infusions/TPN address hypoaminoacidemia and can dramatically improve NME even before tumor removal
Prognosis
| Setting | Survival |
|---|---|
| Resected, no metastases | ~100% 10-year survival |
| Resected, metastatic disease | ~52% survival (large series of 87 patients) |
| Metastatic, unresected | Guarded; SSAs and chemotherapy provide palliation |
Key Differentials for NME Rash
Since NME is not specific to glucagonoma, the following must be excluded:
- Zinc deficiency (acrodermatitis enteropathica)
- Myelodysplastic syndrome
- Short bowel syndrome
- Hepatic cirrhosis
- Celiac disease / malabsorption states
- Inflammatory bowel disease
- Nutritional deficiency states
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025), Ch. 89 and MEN1 chapter
- Yamada's Textbook of Gastroenterology, 7th ed., pp. 1773-1778
- Sabiston Textbook of Surgery, 21st ed., p. 1594
Somatostatinoma
Finding Sources
Searching PubMed
somatostatinoma pancreatic neuroendocrine tumor
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Excellent - I now have comprehensive information from four major textbooks. Here is the full write-up:
Somatostatinoma
Somatostatinoma is an extremely rare neuroendocrine tumor (NET) arising from somatostatin-secreting D cells (delta cells), located most commonly in the pancreas or duodenum. Its incidence is approximately 1 in 40 million - even rarer than glucagonoma. First described in 1977, only a few hundred cases exist in the literature.
Pathophysiology
Somatostatin is a broad-spectrum inhibitory hormone. Excess somatostatin from the tumor suppresses virtually every GI function:
| Target | Effect of Excess Somatostatin | Clinical Consequence |
|---|---|---|
| Insulin | Inhibits secretion | Diabetes mellitus |
| Cholecystokinin (CCK) | Inhibits release + gallbladder contraction | Cholelithiasis |
| Pancreatic enzymes & bicarbonate | Inhibits secretion | Steatorrhea / malabsorption |
| Gastric acid | Inhibits secretion | Hypochlorhydria |
| Intestinal absorption | Inhibits amino acid, sugar, Ca²⁺ absorption | Weight loss / malnutrition |
| Glucagon, gastrin, secretin | Inhibited | Varied GI effects |
Classic Triad ("Somatostatinoma Syndrome")
Diabetes + Cholelithiasis + Diarrhea/Steatorrhea
This triad is classically seen with pancreatic tumors. Duodenal somatostatinomas rarely cause the full hormonal syndrome (only ~3% vs. ~19% of pancreatic tumors).
Clinical Features
| Feature | Detail |
|---|---|
| Diabetes mellitus | >33% of patients; ranges from oral hypoglycemic-controlled to insulin-requiring |
| Cholelithiasis | 16-64% of patients; due to inhibited CCK and gallbladder contraction |
| Steatorrhea / diarrhea | Due to inhibited pancreatic enzymes, bile, and lipid absorption |
| Weight loss | 20-30%; due to malabsorption |
| Hypochlorhydria | From inhibited gastric acid secretion |
| Anemia | Mild; commonly present |
| Jaundice / biliary obstruction | Particularly with periampullary or duodenal tumors |
| Abdominal pain, GI bleeding | More typical of duodenal tumors growing locally |
Key Point - Pancreatic vs. Duodenal Presentation
| Feature | Pancreatic | Duodenal |
|---|---|---|
| Hormonal syndrome | ~19% | ~3% |
| Typical size | 5-6 cm | ~2 cm |
| Psammoma bodies on histology | Rare | Characteristic |
| Symptoms at presentation | Hormonal (triad) | Local mass effect (pain, bleeding, obstruction) |
| Association with NF1 | 6-21% | 43% |
| Periampullary location | Less common | 20% |
Epidemiology & Tumor Characteristics
- Age: 40-70 years; equal sex distribution
- Usually solitary - multiple tumors suggest MEN1
- Located equally in pancreas and GI tract; most common sites: duodenum > pancreatic head > pancreatic tail
- Pancreatic tumors: mean size 5-6 cm (large at presentation)
- Duodenal tumors: mean size ~2 cm
- Metastatic at presentation in 37-90% (liver, lymph nodes, bone); metastatic risk correlates with tumor size
- Strong association with NF1 (von Recklinghausen's disease) - especially duodenal/ampullary tumors
- Rarely associated with MEN1 (unlike other functional PNETs)
Histological Hallmark
Duodenal somatostatinomas characteristically show psammoma bodies on histology - a distinguishing feature not seen in pancreatic somatostatinomas or most other GI NETs.
Diagnosis
Most somatostatinomas are discovered incidentally on imaging or during surgery for another condition, because the symptoms are insidious and nonspecific.
Active suspicion when the classic triad (diabetes + gallstones + steatorrhea) is present, or when an ampullary/periduodenal mass is found in a patient with NF1.
Diagnostic confirmation:
- Elevated fasting serum somatostatin level - this is the definitive test
- Many NETs stain positive for somatostatin on immunohistochemistry (IHC) without causing the syndrome; therefore, elevated serum levels + clinical symptoms are both required to diagnose true somatostatinoma syndrome
- Provocative testing (arginine, tolbutamide stimulation) does not aid in diagnosis
Imaging: CT/MRI for localization and staging; Ga-68 DOTATATE PET-CT for functional imaging and metastatic assessment.
Treatment
Medical
- Nutrition: Malnutrition from steatorrhea can be severe - may require hyperalimentation/TPN
- Diabetes management: Oral hypoglycemics or insulin depending on severity
- Paradox: Somatostatin analogues (octreotide, lanreotide) have shown some benefit in reducing somatostatin release and mitigating diarrhea and diabetes, despite acting on the same receptor - the benefit is thought to come from preferential receptor subtype binding
- Treatment of cholelithiasis as needed
Surgical
- Surgery is recommended for all patients with localized disease - can be curative
- For NF1-associated periampullary somatostatinomas presenting as smaller, localized tumors, surgery is particularly effective
- The potential role of SSAs as primary treatment is questionable given that the tumor itself produces somatostatin
Metastatic Disease
- Streptozotocin + 5-fluorouracil (chemotherapy)
- Hepatic artery embolization for liver metastases
- Everolimus or sunitinib for advanced PNETs
- Peptide receptor radionuclide therapy (PRRT) with Lu-177 DOTATATE for somatostatin receptor-positive disease
Key Distinguishing Points vs. Other Functional PNETs
| Feature | Somatostatinoma | Glucagonoma | VIPoma | Insulinoma |
|---|---|---|---|---|
| Classic triad | DM + gallstones + steatorrhea | NME rash + DM + weight loss | Watery diarrhea + hypokalemia + achlorhydria | Hypoglycemia (Whipple's triad) |
| Key hormone | Somatostatin | Glucagon | VIP | Insulin |
| Location | Pancreas / duodenum | Pancreas (tail) | Pancreas (tail) | Pancreas |
| MEN1 association | Rare | ~10% | Occasional | ~10% |
| NF1 association | Strong (duodenal) | No | No | No |
| Histology clue | Psammoma bodies (duodenal) | - | - | - |
| Metastatic at presentation | 37-90% | 42-78% | Less common | <10% |
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025)
- Yamada's Textbook of Gastroenterology, 7th ed., pp. 1774-1778
- Sabiston Textbook of Surgery, 21st ed., p. 1594
- Fischer's Mastery of Surgery, 8th ed.
Give more detailed answer
Finding Sources
Searching PubMed
somatostatinoma diagnosis treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have rich, multi-source information. Here is the detailed comprehensive answer:
Somatostatinoma - Detailed Review
Overview & Historical Background
Somatostatinoma is a neuroendocrine neoplasm arising from somatostatin-secreting D (delta) cells found in the pancreatic islets, gastric/duodenal/intestinal mucosa, and other sites. The tumor ectopically secretes somatostatin in excess, producing a clinical syndrome first described in 1977 (Krejs et al., NEJM). It is the rarest of the well-characterized functional PNETs, with an annual incidence of approximately 1 in 40 million individuals. Only a few hundred cases exist in the published literature.
Physiology of Somatostatin - Why the Syndrome Makes Sense
Somatostatin is a 14- or 28-amino acid cyclic peptide secreted by D cells in the stomach, duodenum, and pancreatic islets. Under physiological conditions, it acts as a broad-spectrum inhibitor of GI function:
| Somatostatin Inhibits | Consequence of Excess |
|---|---|
| Insulin secretion (pancreatic beta cells) | Diabetes mellitus |
| Cholecystokinin (CCK) release + gallbladder contraction | Cholelithiasis (bile stasis) |
| Pancreatic enzyme & bicarbonate secretion | Steatorrhea, maldigestion |
| Gastric acid secretion | Hypochlorhydria |
| Intestinal absorption of amino acids, sugars, calcium | Malabsorption, weight loss |
| Glucagon, gastrin, secretin release | Multiple metabolic effects |
| Biliary secretion | Contributes to gallstone formation |
The combination of these inhibitory effects forms the basis of the somatostatinoma syndrome.
Anatomical Location
Somatostatinomas are found equally in the pancreas and the GI tract. The most frequent sites:
- Duodenum (most common GI site, often periampullary)
- Pancreatic head (60-80% of pancreatic somatostatinomas occur here)
- Pancreatic tail
- Rarely: jejunum, other intestinal sites
20% are periampullary and many of these have a more aggressive local course (biliary obstruction, jaundice).
Key Clinical Features
The Classic Triad (Somatostatinoma Syndrome)
Diabetes Mellitus + Cholelithiasis + Diarrhea/Steatorrhea
However, this full triad is only commonly seen with pancreatic somatostatinomas. Many tumors - particularly duodenal ones - do not cause the complete syndrome. Most somatostatinomas are diagnosed incidentally.
Detailed Symptom Profile
1. Diabetes Mellitus
- Develops in over 1/3 of patients due to somatostatin's inhibition of insulin secretion
- Severity varies widely: mild glucose intolerance controlled by oral hypoglycemics at one end, insulin-dependent diabetes at the other
- Usually milder than type 1 DM because glucagon secretion is simultaneously inhibited (reducing the counter-regulatory hyperglycemic response)
2. Cholelithiasis / Gallbladder Disease
- Present in 16-64% of patients
- Due to two mechanisms: (a) inhibition of CCK release, and (b) direct inhibition of gallbladder contractility
- Results in bile stasis and gallstone formation
- Right upper quadrant pain is a common presenting complaint (25% of cases, per Schwartz's)
- Jaundice occurs in ~25% - from either gallstone disease or direct periampullary tumor compression of the bile duct
3. Diarrhea & Steatorrhea
- Due to multiple mechanisms: inhibited pancreatic enzyme secretion, inhibited bicarbonate output, inhibited gallbladder emptying, reduced lipid absorption
- Can range from loose stools to florid steatorrhea with bulky, fatty, malodorous stools
- Contributes to significant weight loss
4. Weight Loss
- Present in 20-30% of cases
- Multifactorial: malabsorption, steatorrhea, reduced nutrient absorption
5. Hypochlorhydria
- From inhibited gastric acid secretion; usually subclinical
6. Mild Anemia
- Commonly present; mechanism likely related to malabsorption and nutritional deficiency
7. Abdominal Pain
- The most common presenting complaint in surgical series (25%)
- Can be from cholelithiasis, biliary obstruction, or local tumor mass effect
8. Jaundice
- In ~25% at presentation
- Particularly with periampullary or large pancreatic head tumors compressing the bile duct
Tumor Biology & Histopathology
Pancreatic vs. Duodenal Somatostatinomas - Key Differences
| Feature | Pancreatic | Duodenal/Intestinal |
|---|---|---|
| Causes clinical syndrome | ~19% | ~3% |
| Mean tumor size | 5-6 cm | 2-2.4 cm |
| Psammoma bodies on histology | Rare / absent | Present in ~11-61% |
| Metastases at diagnosis | 70-90% | 30-40% |
| Location | Head (60-80%) | Periampullary (20%+) |
| NF1 association | 6-21% | 43% (duodenal most common tumor in NF1 periampullary region) |
| Behavior | More aggressive | Less malignant, better prognosis |
Psammoma Bodies
These are laminated calcified concentric whorls seen on histology. Their presence in a duodenal NET is a strong clue pointing toward somatostatinoma, even if serum somatostatin is not dramatically elevated. Note: immunostaining positive for somatostatin alone (without elevated serum levels and clinical symptoms) does NOT equal somatostatinoma syndrome.
Size & Malignancy
- Pancreatic tumors are almost always large at diagnosis (mean 5-6 cm)
- Tumor size is the strongest predictor of metastatic spread
- Metastases occur to: liver, lymph nodes, bone
- Overall metastatic rate at presentation: 37-90%
Genetic Associations
1. Neurofibromatosis Type 1 (NF1 / von Recklinghausen's Disease)
- The strongest genetic association for somatostatinoma
- ~1% of NF1 patients develop duodenal NETs, the majority periampullary
- In a 20-year review of periampullary/duodenal neoplasms in NF1 (Relles et al., 2010): somatostatinoma was the most common tumor type at 40%, followed by GIST (34%), adenocarcinoma (8%), carcinoid (6%)
- NF1-associated somatostatinomas:
- Usually periampullary/duodenal (not pancreatic)
- Smaller (mean 2.8 cm vs. 5.9 cm)
- Lower rate of metastases (30% vs. 71%)
- High rate of psammoma bodies (61% vs. 0%)
- Rarely cause the full somatostatinoma syndrome - come to attention due to biliary obstruction, pancreatitis, or bleeding
- Behave more like sporadic duodenal somatostatinomas
2. MEN1 (Multiple Endocrine Neoplasia Type 1)
- Somatostatinomas are only rarely associated with MEN1, in contrast to insulinoma, gastrinoma, and glucagonoma
- Multiple tumors should raise suspicion for MEN1 (somatostatinomas are otherwise almost always solitary)
3. Polycythemia-Paraganglioma/Pheochromocytoma-Somatostatinoma Syndrome
- A rare inherited syndrome, primarily associated with duodenal somatostatinomas
- Associated with VHL or EPAS1 (HIF-2α) mutations
- Should be considered when somatostatinoma co-exists with pheochromocytoma or paraganglioma
Diagnosis
Clinical Suspicion
Somatostatinoma should be suspected when:
- Classic triad (DM + gallstones + steatorrhea) is present together
- Periampullary/duodenal mass found - especially in a patient with NF1
- Psammoma bodies found on biopsy of a duodenal NET
- Any duodenal lesion in an NF1 patient
Biochemical Diagnosis
- Elevated fasting serum somatostatin-like immunoreactivity in plasma - required for diagnosis of the syndrome
- Normal upper limit: typically <120 pg/mL
- Somatostatinoma: usually >500 pg/mL; Schwartz's states levels are usually >10 ng/mL (10,000 pg/mL) in confirmed cases
- Must also confirm elevated levels in the resected tumor tissue
Important caveat: Other tumors outside the pancreas/intestine can also produce somatostatin-like immunoreactivity and should be excluded:
- Small cell lung cancer
- Medullary thyroid carcinoma
- Pheochromocytomas
- Paragangliomas
Many nonfunctional NETs stain positive for somatostatin on IHC without elevated serum levels. These are not somatostatinomas - the diagnosis requires both elevated serum levels and clinical symptoms.
Provocative Testing
- Provocative tests (arginine stimulation, tolbutamide stimulation) do not aid diagnosis and are not recommended
Supporting Labs
- Hyperglycemia (often mild-moderate)
- Hypochlorhydria on gastric pH studies
- Elevated fecal fat (steatorrhea confirmation)
- Imaging: gallstones on ultrasound
Imaging & Localization
- CT abdomen/pelvis with contrast - initial modality; pancreatic head mass or periampullary lesion
- MRI - superior soft tissue detail for pancreatic lesions
- Somatostatin receptor scintigraphy (Octreoscan) or Ga-68 DOTATATE PET-CT - functional imaging to assess somatostatin receptor expression, stage disease, identify metastases; also guides PRRT candidacy
- Endoscopic ultrasound (EUS) - excellent for small duodenal or pancreatic head lesions; allows FNA biopsy
- Upper GI endoscopy - for periampullary and duodenal tumors
Treatment
Surgical (Primary Treatment)
- Surgery is recommended for all patients with localized disease - can be curative
- Attempt at complete excision warranted even in most metastatic cases (debulking)
- Cholecystectomy should be performed at the same time given the high rate of cholelithiasis
- Specific operations:
- Pancreaticoduodenectomy (Whipple) for pancreatic head or periampullary tumors
- Distal pancreatectomy for body/tail pancreatic tumors
- Local duodenal excision for small duodenal tumors (if feasible)
- In NF1-associated periampullary lesions presenting as small, localized tumors, surgery is particularly effective
Medical / Symptom Control
- Somatostatin analogues (SSAs) - octreotide (10-30 mg IM every 4 weeks) or lanreotide (90-120 mg deep SC monthly):
- Paradoxically can improve symptoms by decreasing somatostatin release (via preferential receptor subtype binding)
- Shown to reduce diarrhea and ameliorate diabetes
- Also have antiproliferative effects on NETs
- Diabetes management: oral hypoglycemics or insulin based on severity
- Nutritional support: steatorrhea-related malnutrition can be severe; may require hyperalimentation/TPN, pancreatic enzyme replacement therapy (PERT)
- Pancreatic enzyme replacement for steatorrhea/maldigestion
Management of Metastatic / Unresectable Disease
Similar to other advanced unresectable PNETs:
- SSAs (octreotide LAR, lanreotide) - antiproliferative + symptomatic
- Chemotherapy: streptozotocin + 5-fluorouracil (or streptozotocin + doxorubicin)
- Targeted therapy:
- Everolimus (mTOR inhibitor) - approved for advanced PNETs
- Sunitinib (VEGFR inhibitor) - approved for advanced PNETs
- Peptide Receptor Radionuclide Therapy (PRRT): Lu-177 DOTATATE (Lutathera) - for somatostatin receptor-positive disease; high uptake on DOTATATE PET predicts response
- Liver-directed therapies for hepatic metastases:
- Transarterial embolization (TAE)
- Transarterial chemoembolization (TACE)
- Radioembolization (TARE) with Yttrium-90
- Percutaneous/surgical ablation (radiofrequency or microwave) for oligometastatic disease
- Surgical debulking if >90% tumor burden reducible - 5-year survival 60-75% in selected patients
Important Surgical Note
Since somatostatin inhibits gallbladder function, somatostatin analogues used medically also cause gallstone formation - patients on long-term SSA therapy should be monitored with periodic ultrasound for cholelithiasis.
Prognosis
| Tumor Type | Metastases at Diagnosis | 5-Year Survival |
|---|---|---|
| Intestinal/duodenal somatostatinoma | 30-40% | >80% |
| Pancreatic somatostatinoma | 70-90% | <50% |
| Metastatic PNET (general) with liver mets | - | <50% |
| After surgical debulking (>90% reduction) | Metastatic | 60-75% |
Key prognostic factors:
- Location (duodenal > pancreatic in outcome)
- Tumor size (strongest predictor of metastatic spread)
- Presence of liver metastases at diagnosis
- Resectability of primary tumor
Surveillance post-resection: CT ± somatostatin receptor scan annually for 5 years.
Differential Diagnosis of the Somatostatinoma Syndrome
The triad of DM + gallstones + steatorrhea has many other causes that must be excluded:
| Condition | Overlap Features |
|---|---|
| Chronic pancreatitis | DM + steatorrhea + abdominal pain |
| Type 2 DM + cholelithiasis (coincidental) | DM + gallstones |
| Pancreatic exocrine insufficiency | Steatorrhea + DM (if pancreatic) |
| Celiac disease | Steatorrhea + weight loss |
| Other functional PNETs | Various hormonal syndromes |
Summary - High-Yield Points
- Incidence: 1 in 40 million - the rarest functional PNET
- Classic triad: Diabetes + Cholelithiasis + Steatorrhea
- Mechanism: somatostatin inhibits insulin, CCK, pancreatic enzymes, bile
- Location: pancreas = GI tract (duodenum most common GI site)
- Pancreatic tumors: large (5-6 cm), high metastatic rate (70-90%), cause the full syndrome
- Duodenal tumors: smaller (2 cm), lower metastatic rate (30-40%), psammoma bodies, rarely cause syndrome
- Strong association with NF1 (duodenal/periampullary - most common periampullary tumor in NF1)
- Rarely associated with MEN1
- Diagnosis: elevated fasting serum somatostatin + clinical syndrome + imaging
- Histology clue: psammoma bodies in duodenal tumors
- Treatment: surgery (curative intent) + cholecystectomy; SSAs for symptoms; PRRT/everolimus/sunitinib for advanced disease
- Prognosis: duodenal (5-yr >80%) far better than pancreatic (5-yr <50%)
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025)
- Goldman-Cecil Medicine, International Edition
- Yamada's Textbook of Gastroenterology, 7th ed.
- Sabiston Textbook of Surgery, 21st ed.
- Schwartz's Principles of Surgery, 11th ed.
- Fischer's Mastery of Surgery, 8th ed.
- Sleisenger and Fordtran's GI and Liver Disease
- Mulholland and Greenfield's Surgery, 7th ed.
This is a shared conversation. Sign in to Orris to start your own chat.