Well elaborated, well described pdf on lecture topic screening of urine for inborn error and describe use of paper chromatography for mbbs 1st year Biochemistry students lectures
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Running Command
mkdir -p /home/daytona/workspace/urine-screening-lecture && ls /home/daytona/workspace/
Show me the content don’t generate pdf i’ll read here
Here is the full lecture content for MBBS 1st Year Biochemistry:
Screening of Urine for Inborn Errors of Metabolism & Paper Chromatography
MBBS 1st Year | Biochemistry Lecture Notes
PART 1: INBORN ERRORS OF METABOLISM (IEM)
1.1 Introduction
The term "inborn errors of metabolism" was coined by Sir Archibald Garrod in the early 20th century, based on his classic observation of alkaptonuria - a condition where urine darkened on exposure to air. He proposed that certain diseases result from inherited blocks in metabolic pathways.
Key concept: In IEM, a genetically determined enzyme deficiency causes:
- Accumulation of the substrate (before the block)
- Deficiency of the product (after the block)
- Accumulation of abnormal metabolites (via alternate pathways)
These abnormal metabolites are excreted in urine, making urine an excellent specimen for screening.
1.2 Why Screen Urine?
- Many IEMs cause accumulation of abnormal metabolites that spill into urine
- Urine collection is non-invasive - ideal for neonates and infants
- Early diagnosis allows dietary or therapeutic intervention before irreversible damage (especially brain damage) occurs
- Most IEMs causing aminoacidurias present in the neonatal or early infant period
Two types of aminoaciduria:
| Type | Mechanism | Example |
|---|---|---|
| Overflow type | Enzyme block causes accumulation in blood, then spills into urine | Phenylketonuria (PKU) |
| Renal type | Defect in renal tubular reabsorption; blood levels normal | Cystinuria |
1.3 Common IEMs Detected by Urine Screening
A. Phenylketonuria (PKU)
Enzyme deficient: Phenylalanine hydroxylase (PAH)
Inheritance: Autosomal recessive
Incidence: ~1 in 11,000 births
Metabolic block:
Phenylalanine --[PAH blocked]--> Tyrosine (cannot proceed)
↓ (alternate pathway)
Phenylpyruvic acid → Phenylacetic acid → Phenyllactic acid
Urine findings:
- Elevated phenylpyruvic acid (highest)
- Elevated phenylacetic acid (gives characteristic mousy/musty odor)
- Elevated phenylalanine
- Decreased 5-hydroxyindoleacetic acid
- Elevated urinary indoles (from altered tryptophan metabolism)
Clinical features:
- Mental retardation (if untreated)
- Fair skin and hair (reduced tyrosine → reduced melanin)
- Eczema
- Musty body odor
Urine Screening Tests for PKU:
| Test | Reagent | Positive Result | Notes |
|---|---|---|---|
| Ferric Chloride (FeCl3) test | 10% FeCl3 added to urine | Blue-green color (fades quickly) | Detects phenylpyruvic acid; false positives with salicylates |
| Phenistix strip | Ferric ammonium sulfate + Mg sulfate + cyclohexylsulfamic acid | Grey to grey-green color at 30 sec | Detects 5-10 mg/dL; pink-purple = salicylates/phenothiazines |
| Guthrie Bacterial Inhibition Test | Blood on filter paper, B. subtilis agar + β-thienylalanine inhibitor | Bacterial colony growth = excess phenylalanine | Neonatal blood screen (heel prick) |
Confirmatory test: Ion-exchange HPLC or tandem mass spectrometry
Treatment: Low phenylalanine diet started in first weeks of life
B. Alkaptonuria
Enzyme deficient: Homogentisic acid oxidase
Inheritance: Autosomal recessive
Metabolic block:
Phenylalanine → Tyrosine → p-Hydroxyphenylpyruvate → Homogentisic acid
↓ [BLOCKED]
(accumulates in blood & urine)
Urine findings:
- Large amounts of homogentisic acid in urine
- Urine turns dark brown-black on standing or at alkaline pH (due to oxidation of homogentisate)
Clinical features:
- Dark urine (may be the only early sign)
- Ochronosis - dark-blue to black pigmentation in cartilage and connective tissue (occurs later)
- Arthritis (late complication)
Urine Screening Tests:
| Test | Reagent | Positive Result |
|---|---|---|
| Ferric Chloride test | 2 drops 10% FeCl3 to ~2 mL urine | Transient dark-blue color |
| Silver Nitrate test | 4 mL 3% AgNO3 + 0.5 mL urine + few drops 10% NH4OH | Black color |
| Alkaline urine | NaOH added to urine | Immediate darkening |
Confirmatory test: Paper or thin-layer chromatography, capillary electrophoresis
C. Maple Syrup Urine Disease (MSUD)
Enzyme deficient: Branched-chain alpha-keto acid dehydrogenase complex
Inheritance: Autosomal recessive
Affected amino acids: Leucine, Isoleucine, Valine (branched-chain amino acids - BCAAs)
Metabolic block:
Leucine/Isoleucine/Valine → α-Keto acids --[BLOCKED]--> Cannot be further decarboxylated
↓ (accumulate)
Excreted as keto acids in urine
Urine findings:
- Elevated leucine, isoleucine, valine
- Corresponding keto acids elevated in urine
- Urine (and cerumen/ear wax) has characteristic maple syrup / caramelized sugar odor
Clinical features:
- Severe neonatal vomiting, seizures, lethargy, stupor
- Irregular respirations
- Hypoglycemia
- Rapidly comatose and fatal if untreated
Urine Screening Tests:
| Test | Reagent | Positive Result | Notes |
|---|---|---|---|
| 2,4-DNPH test (Dinitrophenylhydrazine) | 10 drops DNPH reagent (100 mg DNPH in 100 mL 2N HCl) to 1 mL clear urine; wait 10 min | Yellow or chalky-white precipitate (insoluble hydrazones) | Detects α-keto acids; also positive in PKU, histidinemia; rule out simple ketonuria first |
Confirmatory test: GC-MS or TLC of urine, NMR spectroscopy
Treatment: Dietary restriction of branched-chain amino acids
D. Tyrosinemia
Types:
- Type I (Tyrosinosis): Deficiency of fumarylacetoacetate hydrolase → succinylacetone accumulates → liver failure, renal dysfunction, rickets
- Type II (Richner-Hanhart syndrome): Deficiency of tyrosine aminotransferase → corneal erosions, palmar/plantar hyperkeratosis, mental retardation
- Neonatal tyrosinemia: Benign, transient; lowered p-hydroxyphenylpyruvate hydroxylase activity; common in premature infants
Urine findings:
- Tyrosine, p-hydroxyphenylacetic acid, p-hydroxyphenylpyruvic acid excreted
- Tyrosine crystals: fine, silky, brown-to-black; precipitate at acid pH; soluble in alkali
Urine Screening Test:
- Nitrosonaphthol test: Nonspecific screening; tyrosine and tyramine form soluble red complexes
- Confirm by chromatography or quantitative serum tyrosine assay
E. Cystinuria
Defect: Defective transport of cystine by renal tubular epithelial cells and gut (not an enzyme deficiency - a transporter defect)
Inheritance: Autosomal recessive
Incidence: ~1 in 10,000 (homozygous)
Amino acids affected: Cystine, Ornithine, Lysine, Arginine (COLA - dibasic amino acids)
Only cystine crystallizes and forms stones (poorly soluble at normal urine pH)
Clinical features:
- Recurrent urinary tract calculi (kidney stones)
- Renal colic, hematuria
Urine findings:
- Colorless, hexagonal crystals of cystine in urine (first morning specimen)
- Massive cystinuria
- Cystine stones are radio-opaque (contain sulfur)
Urine Screening Tests:
| Test | Reagent | Positive Result | Notes |
|---|---|---|---|
| Cyanide-Nitroprusside test (Brand's test) | Sodium cyanide reduces cystine → cysteine; nitroprusside reacts with -SH groups | Red-purple color | Also positive with cysteine, homocystine, ketones |
| Urine microscopy | First morning concentrated urine | Hexagonal cystine crystals | Pathognomonic when found |
F. Hartnup Disease
Defect: Defective renal and intestinal transport of neutral amino acids (especially tryptophan)
Inheritance: Autosomal recessive
Pathophysiology: Reduced tryptophan absorption → reduced nicotinamide (NAD precursor) synthesis → pellagra-like features
Clinical features (the 3 Ds): Dermatitis (photosensitive), Diarrhea, Dementia (cerebellar ataxia in children)
- Aminoaciduria of neutral amino acids without elevated blood levels (renal type)
1.4 Summary Table of IEM Urine Screening Tests
| Disease | Metabolite in Urine | Screening Test | Color/Result |
|---|---|---|---|
| PKU | Phenylpyruvic acid | FeCl3 test | Blue-green |
| PKU | Phenylalanine | Guthrie test / Phenistix | Colony growth / grey-green |
| Alkaptonuria | Homogentisic acid | FeCl3 test | Dark blue |
| Alkaptonuria | Homogentisic acid | Silver nitrate test | Black |
| MSUD | α-Keto acids | DNPH test | Yellow/white precipitate |
| Tyrosinemia | Tyrosine | Nitrosonaphthol test | Red |
| Cystinuria | Cystine | Cyanide-nitroprusside | Red-purple |
| Cystinuria | Cystine crystals | Urine microscopy | Hexagonal crystals |
1.5 Neonatal Screening Programs
The Guthrie Test (historical, first screening program):
- Heel-prick capillary blood sample taken ~7 days after birth (when enzyme expression should be full)
- Blood blotted onto absorbent filter paper (Guthrie card)
- Disk placed on agar seeded with a phenylalanine-requiring strain of Bacillus subtilis + β-thienylalanine (competitive inhibitor of phenylalanine uptake)
- At normal phenylalanine concentrations, bacteria are inhibited and do not grow
- If phenylalanine is elevated (as in PKU), it overcomes the inhibitor and bacteria form visible colonies
Modern approach: Tandem mass spectrometry (MS/MS) has largely superseded the Guthrie test. It can screen for 30+ IEMs from a single dried blood spot simultaneously, including PKU, MSUD, organic acidemias, and fatty acid oxidation disorders.
PART 2: PAPER CHROMATOGRAPHY
2.1 Introduction and Definition
Chromatography is a separation technique based on the differential distribution of compounds between:
- A stationary phase (does not move)
- A mobile phase (moves and carries the sample)
Compounds with stronger affinity for the stationary phase travel more slowly; those with stronger affinity for the mobile phase travel faster. This differential migration achieves separation.
Paper chromatography is a type of planar (partition) chromatography in which:
- Stationary phase: A layer of water (or polar solvent) adsorbed onto cellulose paper fibers
- Mobile phase: An organic solvent system that moves through the paper by capillary action
- The mechanism is primarily partition (distribution between two liquid phases)
2.2 Principle
The sample is spotted onto the paper. As the mobile phase (solvent) moves up (ascending) or down (descending) through the paper, each component partitions between the water bound to the cellulose fibers (stationary phase) and the moving organic solvent (mobile phase).
- A compound that is more polar stays in the stationary aqueous phase → moves shorter distance
- A compound that is less polar moves with the organic mobile phase → moves longer distance
The degree of separation depends on the partition coefficient of each compound between the two phases.
2.3 Rf Value (Retardation Factor)
The key parameter in paper chromatography:
$$R_f = \frac{\text{Distance traveled by the compound (Ds)}}{\text{Distance traveled by the solvent front (Dt)}}$$
Key points about Rf:
- Always a value between 0 and 1 (dimensionless)
- Characteristic for each compound under defined conditions (solvent, paper, temperature)
- Used for identification by comparing unknown Rf with known standard run on the same plate
- If Rf values match AND color with detection reagent matches → compound is identified
- If Rf does not match → compounds are different
Example for amino acids:
| Amino Acid | Approximate Rf (n-butanol:acetic acid:water) |
|---|---|
| Glycine | 0.26 |
| Alanine | 0.38 |
| Phenylalanine | 0.68 |
| Leucine | 0.73 |
2.4 Procedure for Paper Chromatography of Urine Amino Acids
Materials required:
- Whatman No. 1 filter paper
- Capillary tube or micropipette
- Chromatography tank/chamber
- Solvent system (mobile phase)
- Ninhydrin spray reagent (for detection of amino acids)
- Reference standards
Solvent systems commonly used:
- Ascending: n-Butanol : Acetic acid : Water (4:1:5 upper phase) - most common
- Descending: Phenol : Water (80:20)
- 2D chromatography: Two different solvents run at 90° to each other (for complex mixtures)
Step-by-step procedure:
Step 1 - Preparation of paper:
- Mark a pencil baseline 2-3 cm from the bottom of the Whatman paper
- Mark spots at regular intervals (2 cm apart) on the baseline
Step 2 - Sample application:
- Apply a small, concentrated spot (~1-2 µL) of urine (or urine concentrate) using a capillary tube
- Apply known amino acid standards in adjacent lanes
- Allow to dry completely (repeat for concentration if needed)
- Keep spots small (2-3 mm diameter) for good resolution
Step 3 - Development (ascending method):
- Pour solvent into the chromatography tank; allow tank to become saturated with solvent vapors (close lid, wait 30 min)
- Dip the bottom edge of the paper into the solvent (the baseline/sample must be above solvent level)
- Allow solvent to rise by capillary action until it reaches ~2 cm from the top
- Remove the paper; mark the solvent front immediately
Step 4 - Drying:
- Allow the paper to air-dry in a fume hood (organic solvents must evaporate completely)
Step 5 - Detection (Visualization):
- Spray uniformly with 0.2% ninhydrin in acetone
- Heat in an oven at 100°C for 5-10 minutes
- Amino acids react with ninhydrin to give purple/violet spots (Ruhemann's purple)
- Exception: Proline and hydroxyproline give a yellow color with ninhydrin
Step 6 - Calculation:
- Measure the distance from baseline to the center of each spot (Ds)
- Measure the distance from baseline to the solvent front (Dt)
- Calculate Rf = Ds / Dt
- Compare with standards
2.5 Detection Reagents Used in Paper Chromatography
| Reagent | Detects | Color Produced |
|---|---|---|
| Ninhydrin | Amino acids (primary amines) | Purple/violet (Ruhemann's purple); proline → yellow |
| FeCl3 | Phenolic compounds, keto acids | Various colors (blue-green for phenylpyruvate) |
| DNPH | Keto acids / aldehydes | Yellow precipitate |
| Iodine vapor | Lipids, some organic compounds | Brown/yellow spots |
| UV light (254 nm) | Aromatic compounds | Dark spots on fluorescent background |
| Ninhydrin + collidine | Individual amino acids (2D) | Different colors per amino acid |
2.6 Application of Paper Chromatography in Urine IEM Screening
This is the direct clinical application linking both parts of this lecture:
-
Aminoaciduria detection: Paper chromatography of urine separates all amino acids present. In PKU, an excess phenylalanine spot and phenylpyruvate spot are visible. In MSUD, leucine, isoleucine, and valine spots are prominent.
-
Confirmatory diagnosis: After a positive chemical screening test (e.g., FeCl3 positive), paper chromatography confirms which exact amino acid or organic acid is elevated.
-
Monitoring treatment: Dietary restriction therapy in PKU or MSUD can be monitored by comparing amino acid patterns over time.
-
2D paper chromatography: For complex urine samples with multiple aminoacidurias, the paper is developed in one direction, dried, rotated 90°, and developed again in a different solvent - giving a "fingerprint" map of all amino acids.
From Tietz Textbook: "Another application [of planar chromatography] was in the screening of urine for drugs or metabolites such as amino acids that accumulate during hereditary disorders."
From Harper's Illustrated Biochemistry: "In most centers, the bacterial inhibition test has been superseded by chromatographic techniques that permit the detection of a variety of abnormal metabolites, and hence the detection of a variety of different inborn errors of metabolism."
2.7 Types of Chromatography: Comparison (for MBBS context)
| Feature | Paper Chromatography | Thin Layer Chromatography (TLC) | Column Chromatography |
|---|---|---|---|
| Stationary phase | Water on cellulose paper | Silica/alumina on glass/plastic | Silica/ion-exchange resin in column |
| Mobile phase | Organic solvent | Organic solvent | Liquid solvent |
| Mechanism | Partition | Adsorption/partition | Adsorption/ion-exchange |
| Speed | Slow | Faster than paper | Variable |
| Resolution | Moderate | Better than paper | High |
| Cost | Very cheap | Cheap | Moderate-expensive |
| Quantification | Difficult | Difficult | Easy (with detector) |
| Use in lab | Qualitative; amino acids | Qualitative; lipids, drugs | Quantitative separation |
| Retention parameter | Rf value | Rf value | Retention time / volume |
2.8 Advantages and Limitations of Paper Chromatography
Advantages:
- Simple, inexpensive, no sophisticated equipment needed
- Multiple samples can be run simultaneously on the same paper
- Non-destructive (spot can be cut out for further analysis)
- Good for qualitative identification in resource-limited settings
Limitations:
- Primarily qualitative; not precise for quantitative measurement
- Slow (several hours for development)
- Requires careful technique (spot size, tank saturation)
- Low sensitivity compared to HPLC or MS/MS
- Currently being replaced in clinical labs by HPLC and tandem mass spectrometry
PART 3: INTEGRATION - CLINICAL APPROACH TO A SUSPECTED IEM
3.1 Clinical Approach
When an IEM is suspected in a neonate or infant (based on vomiting, seizures, unusual odor, developmental delay, or family history):
Step 1 - Urine physical examination:
- Observe color (dark urine → alkaptonuria)
- Smell (mousy → PKU; maple syrup → MSUD; sweaty feet → isovaleric acidemia)
Step 2 - Urine chemical screening:
- FeCl3 test
- DNPH test
- Cyanide-nitroprusside test
- Ninhydrin test
Step 3 - Chromatographic analysis:
- Paper chromatography or TLC of urine for amino acid pattern
- Ninhydrin staining for identification and Rf comparison
Step 4 - Confirmatory tests:
- Quantitative plasma amino acids by HPLC
- Tandem mass spectrometry (MS/MS)
- Enzyme assay (gold standard)
- Molecular genetics / DNA analysis
PART 4: KEY EXAMINATION POINTS (MBBS 1st Year)
Must-Know Facts
- Garrod coined the term "inborn errors of metabolism" - alkaptonuria was his model disease
- PKU = phenylalanine hydroxylase deficiency; mousy odor; FeCl3 → blue-green; treat with low-Phe diet
- Alkaptonuria = homogentisic acid oxidase deficiency; urine turns dark on standing; ochronosis
- MSUD = branched-chain keto acid dehydrogenase deficiency; maple syrup odor; DNPH test positive
- Cystinuria = renal transport defect (not enzyme deficiency); hexagonal crystals; cyanide-nitroprusside test
- Hartnup disease = neutral amino acid transport defect; pellagra-like (tryptophan deficiency)
- Rf value = distance by compound / distance by solvent front; always 0-1
- Ninhydrin = detection reagent for amino acids → purple color (proline = yellow)
- Overflow aminoaciduria (high blood + high urine) vs Renal aminoaciduria (normal blood + high urine)
- Guthrie test = B. subtilis + β-thienylalanine inhibitor; heel-prick blood at day 7; detects PKU
Likely Exam Questions
Short answer:
- Describe the principle and procedure of paper chromatography for separation of amino acids
- What is the Rf value? How is it calculated?
- Name the urine screening tests for PKU and MSUD with their principle
- Differentiate overflow vs renal type aminoaciduria
Long answer:
- Describe the biochemical basis, urine findings, and screening tests for PKU, MSUD, and alkaptonuria
- Describe paper chromatography with its applications in detection of inborn errors of metabolism
References
- Henry's Clinical Diagnosis and Management by Laboratory Methods, 23rd Ed. - Urinary Screening for Inherited Metabolic Diseases, Chapter on Aminoacidurias
- Tietz Textbook of Laboratory Medicine, 7th Edition - Planar Chromatography (Chapter 19)
- Harper's Illustrated Biochemistry, 32nd Ed. - Screening Neonates for Inborn Errors of Metabolism; Phenylalanine and PKU
Prepared for MBBS 1st Year Biochemistry | Topic: Urine Screening for IEM and Paper Chromatography
Search for clinical images of IEM presentations (ochronosis, etc.)
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
All images retrieved from the textbooks. Here they are, organized by condition:
Clinical Images of IEM Presentations
1. PHENYLKETONURIA (PKU)
Fair skin, light hair + eczematous dermatitis
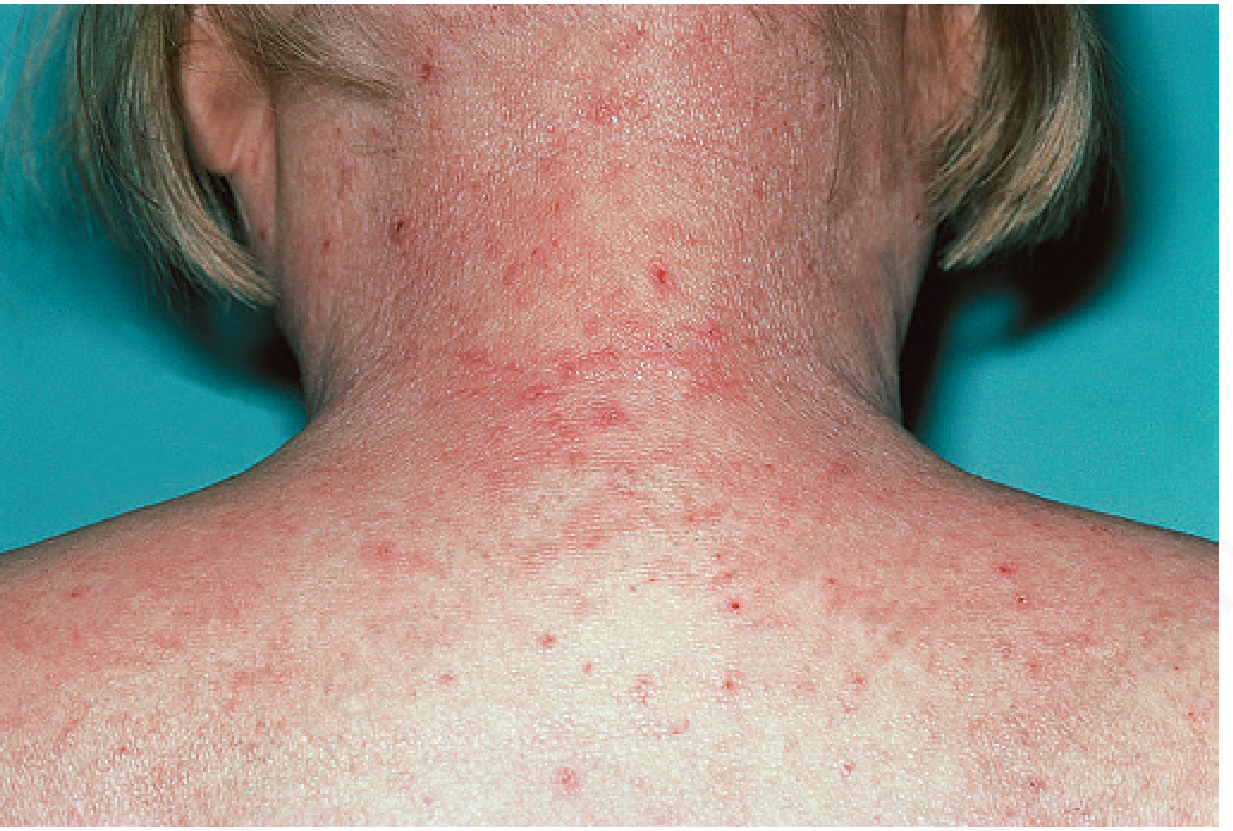
Fig. 26.32 - PKU patient showing characteristic fair skin, light/blonde hair, and eczematous dermatitis over the neck and upper back. Note the scattered red papules - the dermatitis resembles atopic dermatitis and has a predilection for flexures. The patient is described as blond and blue-eyed due to reduced melanin synthesis (tyrosine deficiency → reduced tyrosinase activity).Source: Andrews' Diseases of the Skin - Clinical Dermatology
What to tell students: In PKU, excess phenylalanine competitively inhibits tyrosinase, blocking melanin production. This causes pigmentary dilution - the classic blonde hair and blue eyes - even in families with darker pigmentation.
2. ALKAPTONURIA / OCHRONOSIS
Image A: Osler Sign - Scleral Ochronosis (Conjunctival pigmentation)
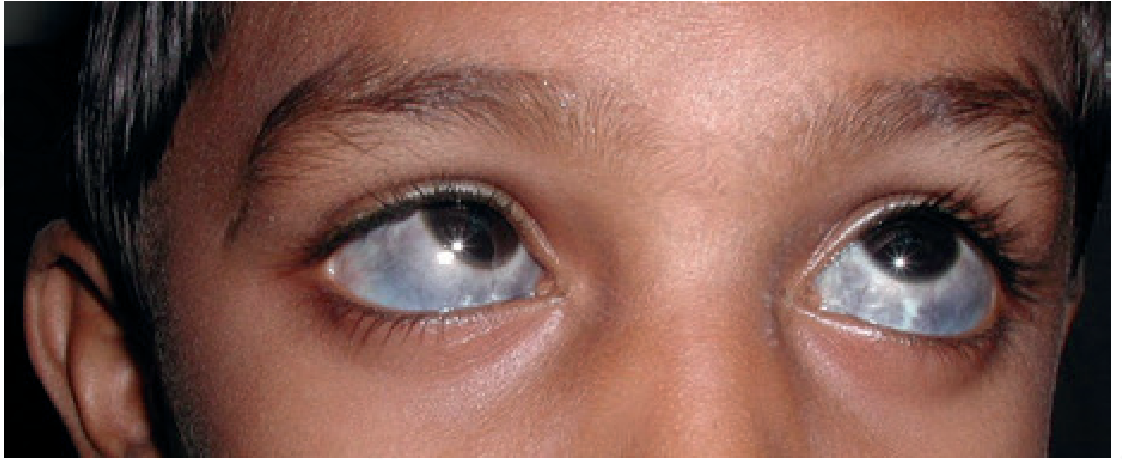
Fig. 26.33 - Osler Sign. Grey-blue/whitish ochronotic pigmentation deposited in the sclera and conjunctiva of both eyes. This is one of the earliest visible signs of alkaptonuria, appearing in the 3rd decade of life as homogentisic acid (HGA) deposits in connective tissues. The pigmentation is caused by oxidation of HGA to benzoquinone acetate which polymerizes and binds collagen.Source: Andrews' Diseases of the Skin - Clinical Dermatology
Image B: Ear Cartilage Ochronosis
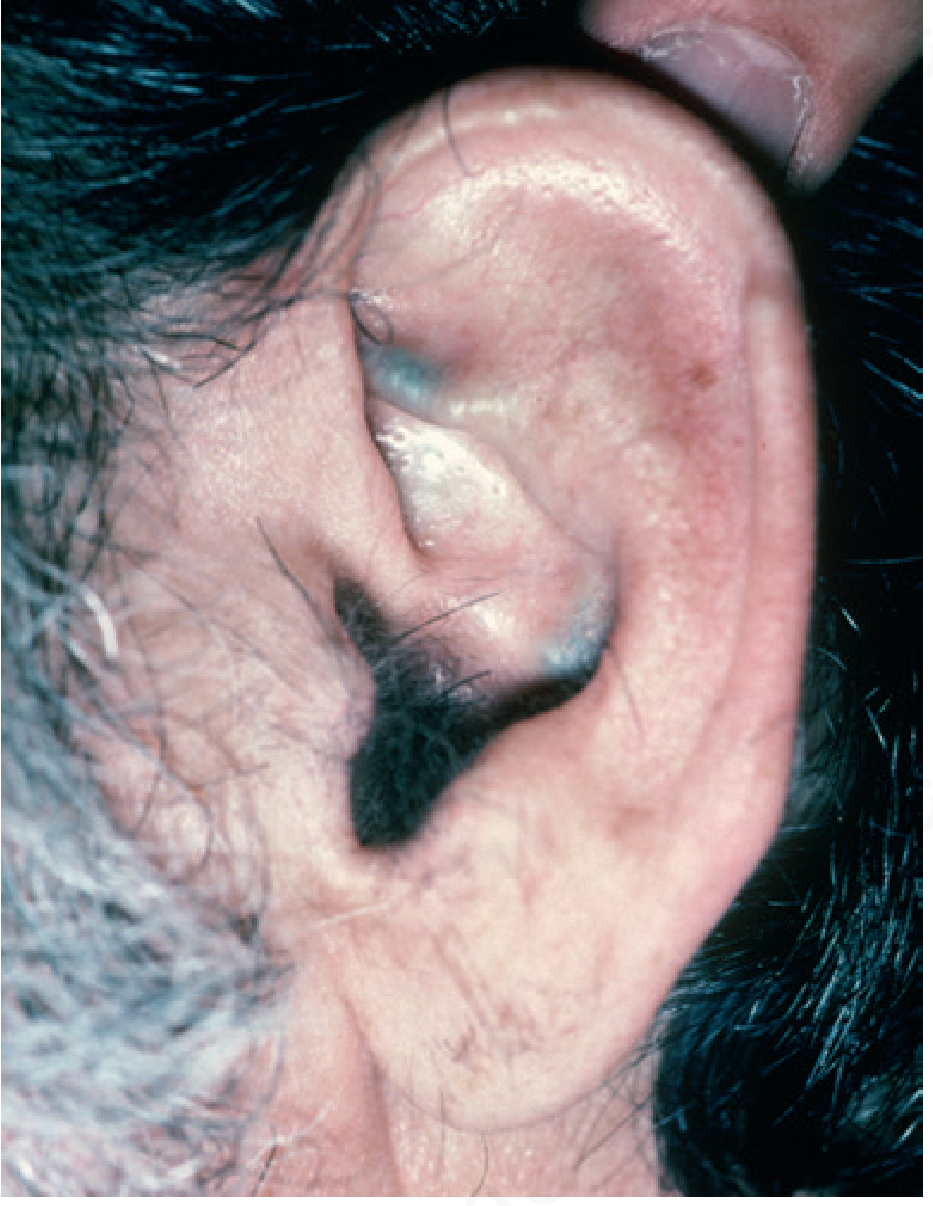
Fig. 26.34 - Alkaptonuria (Ear Ochronosis). Dark blue-grey pigmentation of the auricular (ear) cartilage - one of the earliest and most characteristic signs of ochronosis. The cartilage is rich in collagen, which avidly binds the ochronotic pigment. The ear cartilage and the sclera are the two earliest sites of visible pigmentation.Source: Andrews' Diseases of the Skin - Clinical Dermatology
Image C: Scleral Ochronosis (Ophthalmology view - rectus tendon pigmentation)
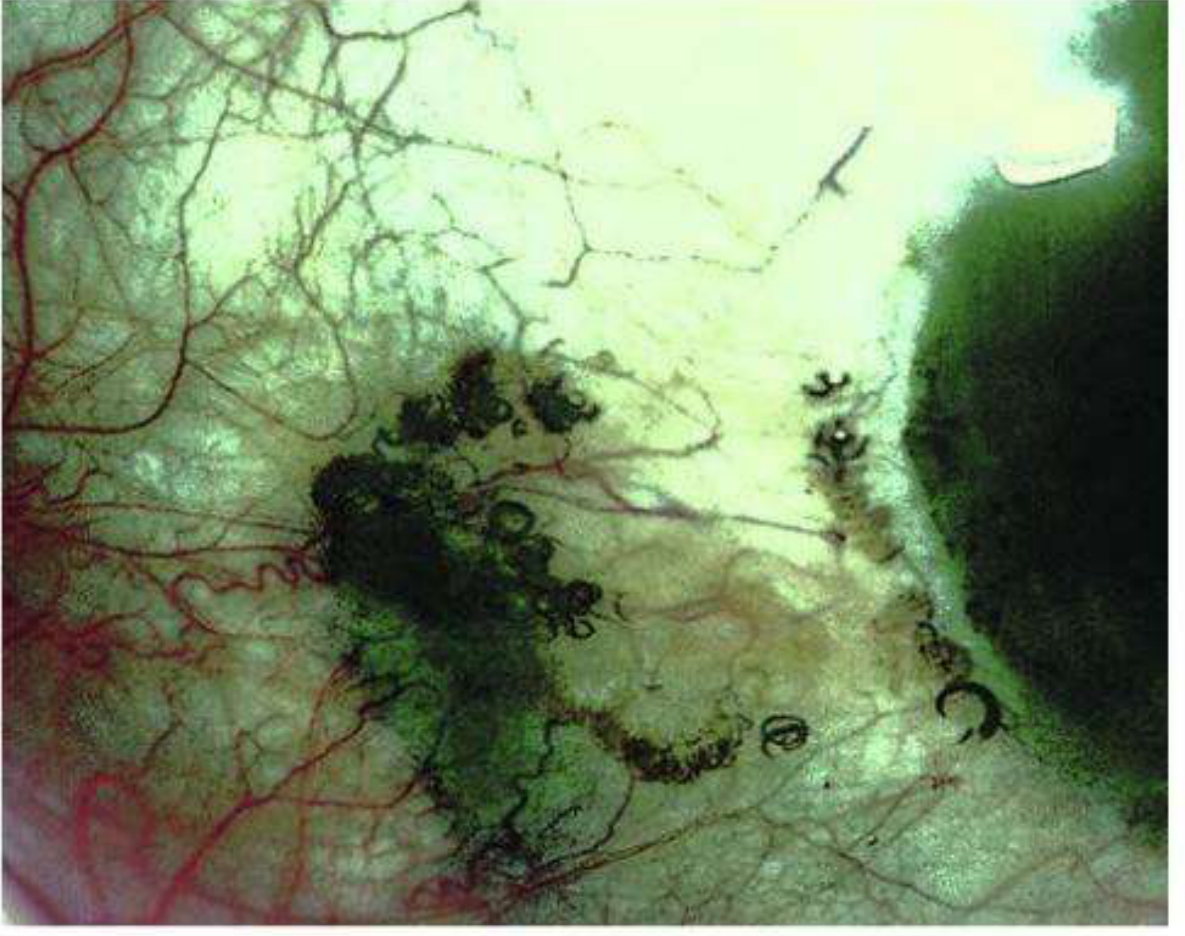
Fig. 9.16 (Kanski's Clinical Ophthalmology) - Alkaptonuria showing heavy dark ochronotic pigmentation of the sclera and the horizontal rectus tendons of the eye. The dark irregular deposits follow the course of the tendons, and the scleral pigmentation is patchy and deep-black. This is a more advanced presentation than the Osler sign above.Source: Kanski's Clinical Ophthalmology, 10th Edition
Progression summary of ochronosis signs:
| Stage | Site | Finding |
|---|---|---|
| Early (3rd decade) | Sclera | Osler sign - grey-blue tint |
| Early-mid | Ear cartilage | Dark blue pigmentation |
| Mid | Nose, tendons, hands | Dark discoloration |
| Late | Spine, large joints | Ochronotic arthropathy, calcified discs |
| Late | Aortic/mitral valves | Cardiac involvement |
3. CYSTINURIA
Three-panel clinical image: Cystine stones + X-ray + Urine microscopy
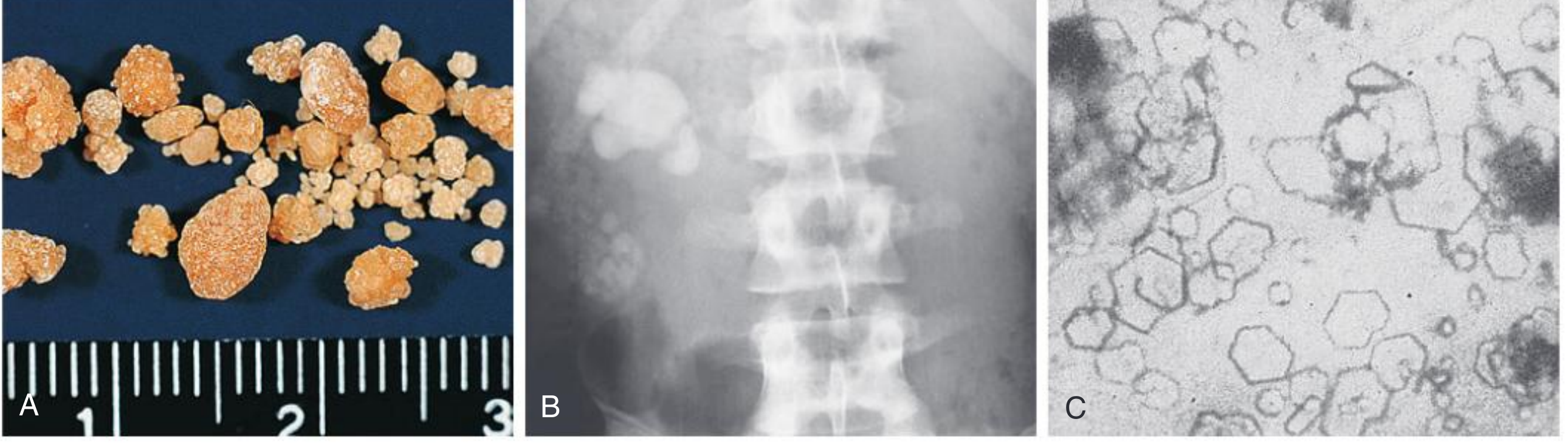
Fig. 50.8 - Cystinuria (Comprehensive Clinical Nephrology)
- Panel A: Rough and smooth cystine calculi (kidney stones) - orange-brown, granular texture, multiple sizes. They are radio-opaque due to sulfur content.
- Panel B: Plain abdominal radiograph showing a cystine calculus in the right renal pelvis and multiple parenchymal calculi.
- Panel C: Urine microscopy - characteristic flat hexagonal crystals of cystine. Their presence is pathognomonic of cystinuria. They are insoluble at urine pH up to 7.4.
Source: Comprehensive Clinical Nephrology, 7th Edition
4. URINE CRYSTALS - MICROSCOPY PANEL (for comparison)

Fig. 4.4 (G-K) - Urinary Crystals - Phase contrast microscopy (Comprehensive Clinical Nephrology)
- Panel G (top left): Cystine crystals - classic hexagonal plates stacked/heaped on one another, with irregular edges and faint rainbow birefringence. These are pathognomonic of cystinuria when found in urine.
- Panels H-K show drug crystals for comparison (2,8-dihydroxyadenine, amoxicillin, ciprofloxacin, indinavir).
Source: Comprehensive Clinical Nephrology, 7th Edition
Quick Visual Summary for Exam
| IEM | What to look for clinically | Pathognomonic finding |
|---|---|---|
| PKU | Fair skin, blonde hair, eczema, musty odor | Dermatitis + pigment dilution + mental delay |
| Alkaptonuria | Grey-blue sclera (Osler sign), dark ear cartilage, dark urine on standing | Ochronosis of sclera + ear cartilage |
| Cystinuria | Recurrent kidney stones, renal colic | Hexagonal crystals in urine microscopy |
| MSUD | Sick neonate, maple syrup odor from cerumen/urine | Odor + DNPH positive |
All images sourced from standard medical textbooks (Andrews' Dermatology, Kanski's Ophthalmology, Comprehensive Clinical Nephrology). These are exactly the clinical images a 1st year MBBS student should be able to recognize in exams and clinical postings.
Search for clinical images of IEM presentations (ochronosis, etc
I already retrieved all those images in the previous response! Here they are again, all together for easy reference:
Clinical Images of IEM Presentations
1. PKU - Fair Skin, Light Hair + Eczematous Dermatitis
What you see: Blonde hair, fair skin, scattered red papules - eczematous/atopic-like dermatitis over neck and upper back. Caused by reduced melanin production (phenylalanine competitively inhibits tyrosinase).
Source: Andrews' Diseases of the Skin
2. Alkaptonuria - Osler Sign (Scleral Ochronosis)
What you see: Grey-white ochronotic pigmentation in the sclera and conjunctiva of both eyes. One of the earliest external signs of alkaptonuria, appearing in the 3rd decade.
Source: Andrews' Diseases of the Skin
3. Alkaptonuria - Ear Cartilage Ochronosis
What you see: Dark blue-grey pigmentation of the auricular cartilage. The ear cartilage (along with sclera) is among the earliest sites of visible ochronosis because cartilage is collagen-rich and avidly binds oxidized homogentisic acid.
Source: Andrews' Diseases of the Skin
4. Alkaptonuria - Ocular Ochronosis + Rectus Tendon Pigmentation (Ophthalmology view)
What you see: Heavy black ochronotic deposits along the sclera and horizontal rectus tendons of the eye. More advanced presentation - deposits follow the tendon course.
Source: Kanski's Clinical Ophthalmology, 10th Ed
5. Cystinuria - Stones + X-Ray + Urine Microscopy (3 panels)
What you see:
- A - Rough, orange-brown cystine kidney stones (radio-opaque due to sulfur)
- B - Plain abdominal X-ray showing cystine calculus in right renal pelvis + parenchymal calculi
- C - Urine microscopy: flat hexagonal crystals - pathognomonic of cystinuria
Source: Comprehensive Clinical Nephrology, 7th Ed
6. Urine Crystal Microscopy - Cystine Crystals (Panel G) vs Others
What you see:
- G (top left) - Cystine crystals: hexagonal plates stacked on each other, faint rainbow birefringence - pathognomonic when found in urine
- H-K - Drug crystals (2,8-dihydroxyadenine, amoxicillin, ciprofloxacin, indinavir) - for comparison
Source: Comprehensive Clinical Nephrology, 7th Ed
Exam Summary
| IEM | Key Clinical Image Finding |
|---|---|
| PKU | Blonde hair + fair skin + eczema |
| Alkaptonuria | Grey sclera (Osler sign) + dark ear cartilage + dark urine |
| Cystinuria | Hexagonal crystals on urine microscopy + radio-opaque stones |
These are all the IEM clinical images available in the textbook library. If you want additional images for MSUD or tyrosinemia, those conditions have limited photographic representations in the available texts since their presentation is mainly neurological/neonatal.
This is a shared conversation. Sign in to Orris to start your own chat.