I'm a second mbbs student. Explain me the pathogenesis of ISCHEMIC HEART DISEASE from pathology which should include: 1. Pathogenesis 2. Chronic vascular occlusion 3. Acute plaque change 4. Consequences of myocardial ischemia The explanation should be on the basis of ROBBINS AND COTRANS PATHOGENESIS OF DISEASES 2ND MBBS use flow charts to explain various topics
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have all the material I need from Robbins & Kumar. Let me now compile a comprehensive, well-organized response with flow charts for the 2nd MBBS student.
Ischemic Heart Disease (IHD) - Pathology
Based on Robbins & Kumar Basic Pathology (Robbins Pathology)
Definition
IHD is a broad term encompassing several closely related syndromes caused by an imbalance between cardiac blood supply (perfusion) and myocardial oxygen/nutritional demand.
In >90% of cases, IHD is a consequence of reduced coronary blood flow secondary to obstructive atherosclerotic vascular disease. Thus, IHD is essentially synonymous with Coronary Artery Disease (CAD).
Clinical Syndromes of IHD
IHD Syndromes
├── Angina Pectoris (ischemia without myocyte death)
│ ├── Stable Angina (predictable with exertion)
│ ├── Prinzmetal/Variant Angina (vasospasm at rest)
│ └── Unstable Angina ──────────┐
├── Myocardial Infarction (MI) ───┤── ACUTE CORONARY
├── Sudden Cardiac Death (SCD) ───┘ SYNDROME (ACS)
└── Chronic IHD with CHF (pump failure)
1. PATHOGENESIS OF IHD
PATHOGENESIS OF IHD
══════════════════════════════════════════════════════
Risk Factors
(HTN, DM, Smoking, Hyperlipidemia, Obesity, Family Hx)
│
▼
ENDOTHELIAL DYSFUNCTION / INJURY
│
▼
Lipid accumulation in intima + Monocyte adhesion
│
▼
Monocytes → Macrophages → Foam Cells (lipid-laden)
│
▼
FATTY STREAK (earliest visible lesion)
│
▼
SMC migration from media → intima
SMC proliferation + ECM elaboration (fibrous cap forms)
Continued lipid + inflammatory cell accumulation
│
▼
FIBROFATTY PLAQUE → ADVANCED/VULNERABLE PLAQUE
(large lipid core + thin fibrous cap)
│
├──────────────────────────────────────┐
│ │
▼ ▼
CHRONIC / FIXED OCCLUSION ACUTE PLAQUE CHANGE
(gradual, stable) (sudden, unstable)
Cardiac myocytes generate energy exclusively through mitochondrial oxidative phosphorylation - they are entirely dependent on continuous oxygenated blood flow. Any reduction causes immediate functional loss.
Two main mechanisms of IHD:
- Preexisting (fixed) atherosclerotic occlusion - chronic, slow progression
- Acute plaque change with superimposed thrombosis and/or vasospasm
2. CHRONIC VASCULAR OCCLUSION
This is the "fixed" obstruction pathway leading to stable, predictable symptoms.
DEGREE OF FIXED LUMINAL OCCLUSION & CONSEQUENCES
═══════════════════════════════════════════════════════════════
< 70% obstruction ──────────────────────→ ASYMPTOMATIC
(no symptoms even with exertion)
> 70% obstruction ──────────────────────→ CRITICAL STENOSIS
(symptoms with exertion) │
▼
STABLE ANGINA
(predictable chest pain
on certain exertion)
≥ 90% obstruction ──────────────────────→ Inadequate flow
even at rest
│
▼
UNSTABLE ANGINA
(rest symptoms)
Slowly progressive occlusion ──→ Collateral vessel formation
(over years) from other coronary arteries
│
▼
Protects against MI
(even if vessel fully occluded)
ACUTE blockage ──────────────────→ NO time for collaterals
│
▼
INFARCTION results
Vessels commonly affected: LAD > RCA > LCX (first several cm from origin for LAD/LCX; entire length for RCA)
Vasospasm and Stimuli:
VASOCONSTRICTION TRIGGERS (worsen critical stenosis)
├── Circulating adrenergic agonists (stress, exercise)
├── Platelet-released products (TXA2, serotonin)
├── Imbalance: ↓ Nitric Oxide / ↑ Endothelin
└── Mediators from perivascular inflammatory cells
3. ACUTE PLAQUE CHANGE
This is the mechanism behind Acute Coronary Syndromes (unstable angina, MI, SCD).
(Fig. 9.7 from Robbins - Diagram of sequential progression):
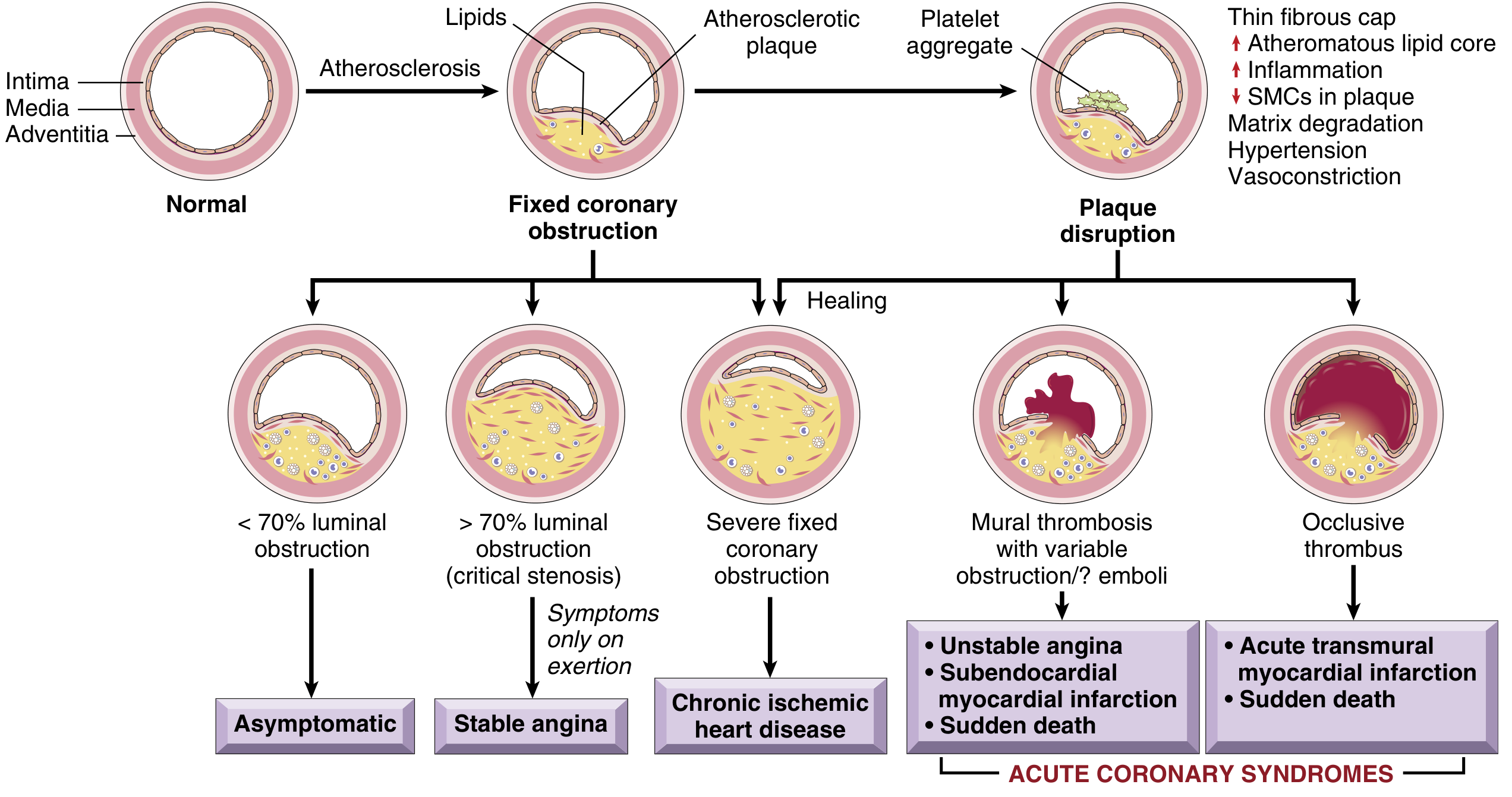
(Natural history of atherosclerosis and acute plaque changes - Fig. 8.12):
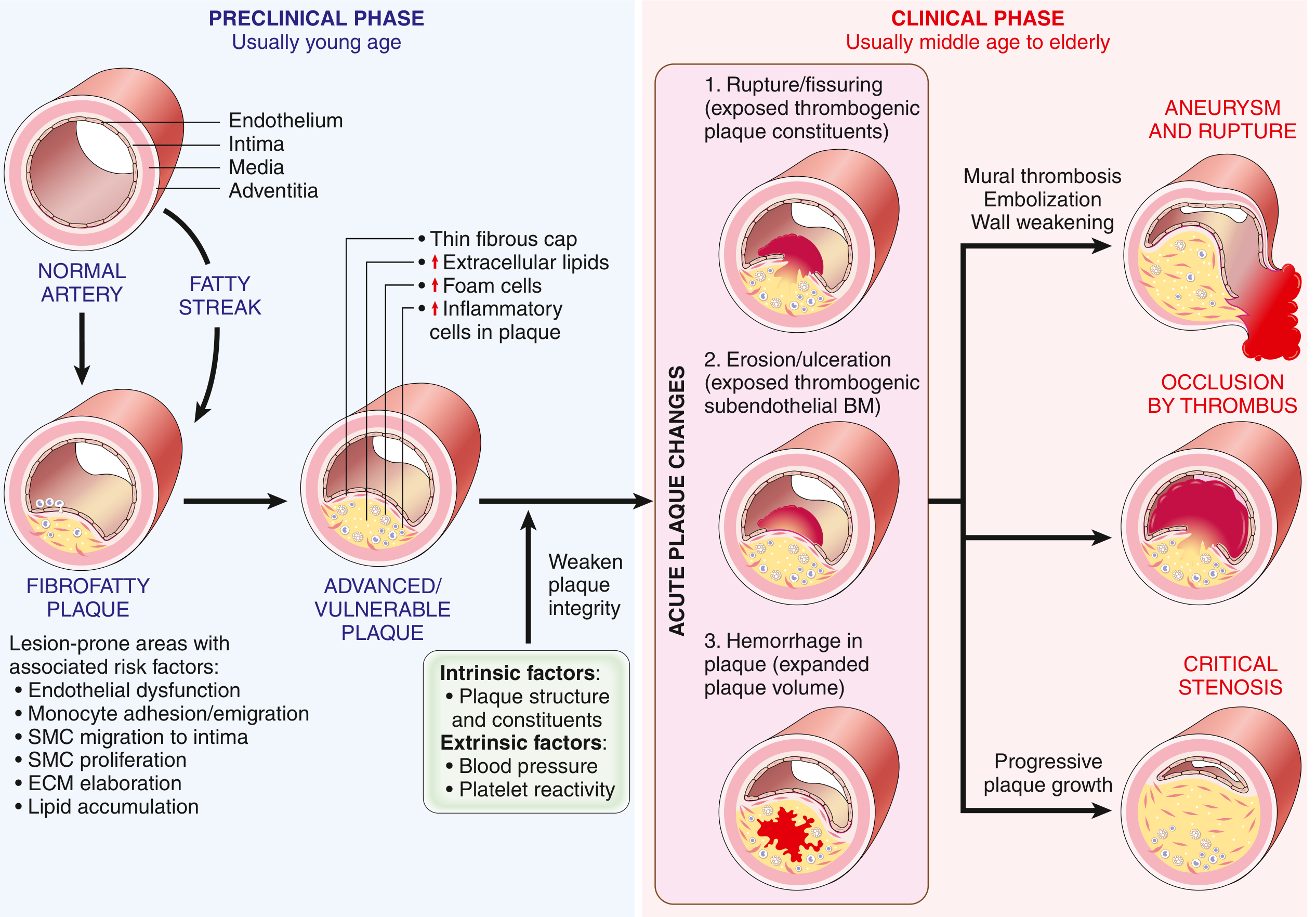
ACUTE PLAQUE CHANGE - MECHANISM
═════════════════════════════════════════════════════
VULNERABLE PLAQUE
(Large lipid core + Thin fibrous cap + many macrophages)
│
├───────────────────────┬──────────────────────┐
│ │ │
▼ ▼ ▼
RUPTURE/FISSURING EROSION/ULCERATION HEMORRHAGE
(mechanical stress) (endothelial apoptosis INTO PLAQUE
at cap-wall junction + inflammatory injury) (neovascularization
ruptures)
│ │ │
▼ ▼ ▼
Exposes thrombogenic Exposes subendothelial Expands plaque
plaque contents basement membrane volume →
(collagen, lipid) Acute luminal
narrowing
└───────────────────────┴──────────────────────┘
│
▼
THROMBUS FORMATION
│
┌─────────────────┴─────────────────┐
│ │
▼ ▼
PARTIAL/MURAL THROMBUS COMPLETE OCCLUSION
± emboli │
│ ▼
▼ TRANSMURAL INFARCTION
Subendocardial infarct (MI)
Unstable angina Sudden Cardiac Death
Key Point on "Vulnerable" Plaques:
- Large atheromatous core + thin fibrous cap = vulnerable
- Cap ruptures at cap-wall junction (max mechanical stress, thinnest cap)
- Macrophages secrete metalloproteases → degrade collagen → weaken cap
- Smooth muscle cells produce collagen → strengthen cap
- Statins help by reducing plaque inflammation and stabilizing plaques
Important: In 2/3 of cases, the culprit plaque causing MI had ≤50% stenosis before rupture - it was NOT critically stenotic or symptomatic! This means many patients have no warning before an acute MI.
Steps in coronary artery occlusion during MI:
1. Plaque erosion/rupture → subendothelial collagen + necrotic
contents exposed to blood
│
▼
2. Platelet adhesion, aggregation, activation
→ Release TXA2, ADP, Serotonin → more platelet aggregation
→ Vasospasm
│
▼
3. Coagulation activated (Tissue Factor exposure)
→ growing thrombus
│
▼
4. Within minutes: complete coronary occlusion
4. CONSEQUENCES OF MYOCARDIAL ISCHEMIA
TIMELINE OF MYOCARDIAL RESPONSE TO ISCHEMIA
═════════════════════════════════════════════════════════
CORONARY OCCLUSION
│
▼ (SECONDS)
Aerobic metabolism ceases
↓ ATP + accumulation of lactic acid
│
▼ (MINUTES - within 1-2 min)
LOSS OF CONTRACTILITY ← earliest functional change
(reversible at this stage)
│
▼ (20-40 MINUTES)
IRREVERSIBLE INJURY begins
Coagulative necrosis of myocytes
Sarcolemmal membrane disruption
→ Leakage of intracellular macromolecules
(Troponin I, Troponin T, CK-MB) → into blood
← BASIS OF CARDIAC BIOMARKERS
│
▼
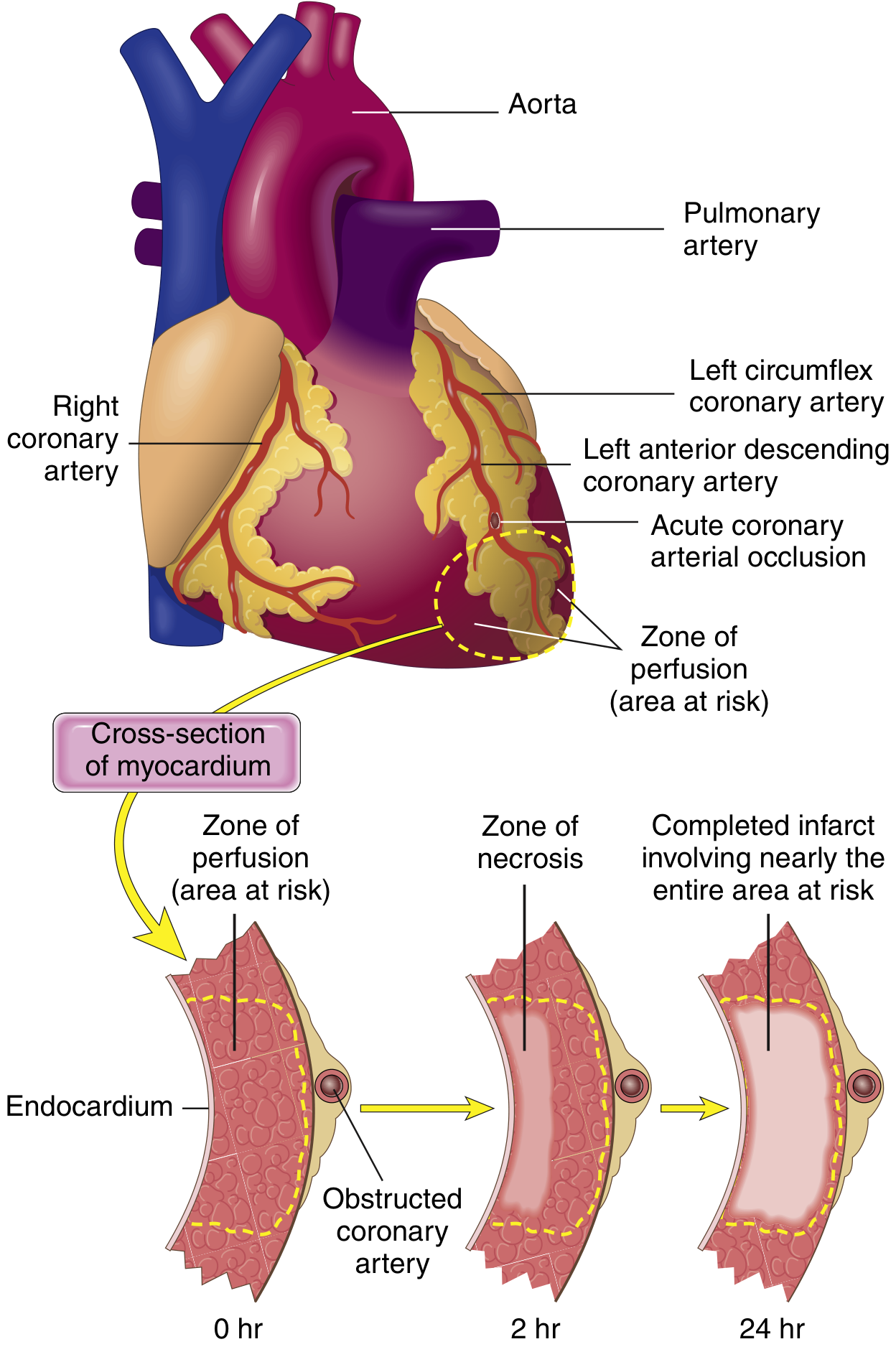
SUBENDOCARDIAL zone affected FIRST
(most susceptible - last to receive blood,
highest intramural pressure)
│
▼ (3-6 HOURS without intervention)
"WAVEFRONT" of necrosis progresses outward
(driven by tissue edema, ROS, inflammatory mediators)
│
▼
TRANSMURAL INFARCT (full wall thickness)
Infarct achieves full extent in 3-6 hours
Morphological Evolution of MI (Table 9.2 - Robbins)
| Time Frame | Gross Features | Light Microscopy |
|---|---|---|
| 0-30 min | None | None (EM: mitochondrial swelling, glycogen loss) |
| 30 min - 4 hr | None | Usually none; waviness of fibers at border |
| 4-12 hr | Occasionally dark mottling | Coagulation necrosis begins; edema; hemorrhage |
| 12-24 hr | Dark mottling | Ongoing coagulative necrosis; pyknotic nuclei; hypereosinophilic myocytes; early neutrophil infiltrate |
| 1-3 days | Mottling with yellow-tan center | Coagulative necrosis; loss of nuclei/striations; increased neutrophils |
| 3-7 days | Hyperemic border; yellow-tan softening | Disintegrating dead fibers; macrophage phagocytosis begins |
| 7-10 days | Max yellow-tan, soft; depressed red-tan margins | Active phagocytosis; early granulation tissue |
| 10-14 days | Red-gray depressed borders | Well-established granulation tissue; new vessels; collagen |
| 2-8 weeks | Gray-white scar (periphery → core) | Increased collagen; decreased cellularity |
| > 2 months | Scar complete | Dense collagenous scar |
Functional Consequences
CONSEQUENCES OF MYOCARDIAL ISCHEMIA
═════════════════════════════════════
ISCHEMIA
│
├─── ARRHYTHMIAS ← electrical instability of ischemic myocardium
│ (80-90% of cardiac deaths in IHD = ventricular fibrillation)
│
├─── STUNNED MYOCARDIUM
│ (reperfused but still non-contractile for days;
│ persistent biochemical abnormalities; reversible)
│
├─── SUBENDOCARDIAL INFARCT
│ (ischemia involving innermost zone only)
│
├─── TRANSMURAL INFARCT
│ (full wall thickness with prolonged occlusion)
│
└─── PUMP FAILURE
(chronic/repeated ischemia → CHF)
Why Subendocardium is Most Vulnerable
SUBENDOCARDIAL VULNERABILITY
├── Most distal from epicardial vessels (last to receive blood)
├── Exposed to highest intramural pressures
│ (impedes blood inflow)
└── Therefore: first to become ischemic, first to necrose
Summary Flowchart - Full IHD Pathogenesis
RISK FACTORS (HTN, DM, Smoking, Hyperlipidemia)
│
▼
Endothelial Dysfunction → Atherosclerosis
│
├──────────────────────────────────┐
│ │
CHRONIC FIXED OCCLUSION ACUTE PLAQUE CHANGE
(gradual over decades) (sudden disruption)
│ │
▼ ▼
< 70%: Asymptomatic Plaque rupture/erosion/hemorrhage
> 70%: Stable Angina │
≥ 90%: Rest symptoms Thrombosis
(Unstable angina) │
│ ┌──────────────────┴──────────────┐
Collaterals ←slow │ │
(protect vs MI) Partial thrombus Complete occlusion
± emboli │
│ ▼
▼ TRANSMURAL MI
Subendocardial MI Sudden Cardiac Death
Unstable angina
│
└─────────────────┐
▼
↓ ATP → Loss of contractility
(seconds-minutes)
│
▼
20-40 min → Irreversible necrosis
(coagulative necrosis)
│
▼
Wavefront subendocardium → transmural
(3-6 hrs for complete infarct)
│
┌──────────────────┴──────────────────┐
│ │ │
Arrhythmia Pump failure Biomarker leak
(VF → SCD) (CHF) (Troponin, CK-MB)
Source: Robbins & Kumar Basic Pathology (Robbins Pathology), 9780323790185, Chapter 9 - Heart Disease
The key takeaway for your exam: IHD results from atherosclerosis causing either a fixed chronic obstruction (stable angina) or acute plaque change with thrombosis (acute coronary syndromes). The 20-40 minute window is critical - before this point, ischemia is reversible; after it, irreversible necrosis sets in. Subendocardium is always the most vulnerable zone due to its position farthest from epicardial blood supply and exposure to high intramural pressures.
This is a shared conversation. Sign in to Orris to start your own chat.