MPGN
Reading File
Finding Sources
Searching PubMed
membranoproliferative glomerulonephritis classification pathogenesis
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have enough high-quality content to write a comprehensive answer. Let me compile everything.
Membranoproliferative Glomerulonephritis (MPGN)
MPGN is not a single disease - it is a histopathological pattern of glomerular injury characterized by mesangial expansion, endocapillary proliferation, GBM thickening, and double contouring ("tram-tracking") of the capillary walls. It accounts for ~10% of kidney biopsies.
Classification (Current Framework)
The old classification into Types I, II, and III (based solely on electron microscopy deposit location) has been replaced by an immunofluorescence (IF)-based system:
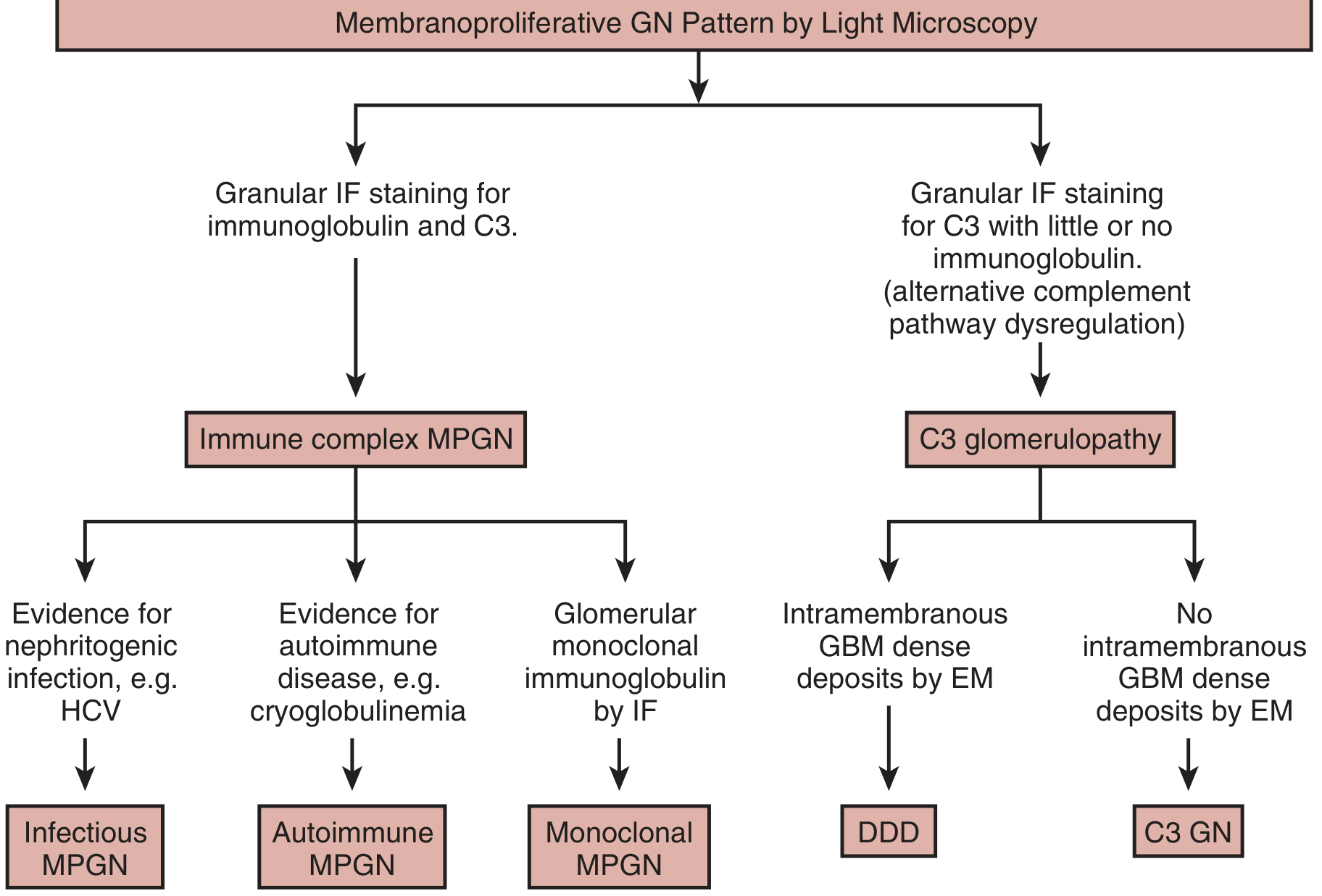
| IF Finding | Category | Further Subclassification |
|---|---|---|
| Ig + C3 (granular) | Immune complex MPGN (IC-MPGN) | Infectious / Autoimmune / Monoclonal |
| C3 dominant (little/no Ig) | C3 Glomerulopathy (C3G) | DDD (intramembranous deposits on EM) / C3 GN (no intramembranous deposits) |
| No Ig, no complement | Consider TMA, malignant hypertension, antiphospholipid syndrome | - |
- Brenner and Rector's The Kidney, p. 1370
- Harrison's Principles of Internal Medicine 22E, p. 2464
Glomerular Schematic
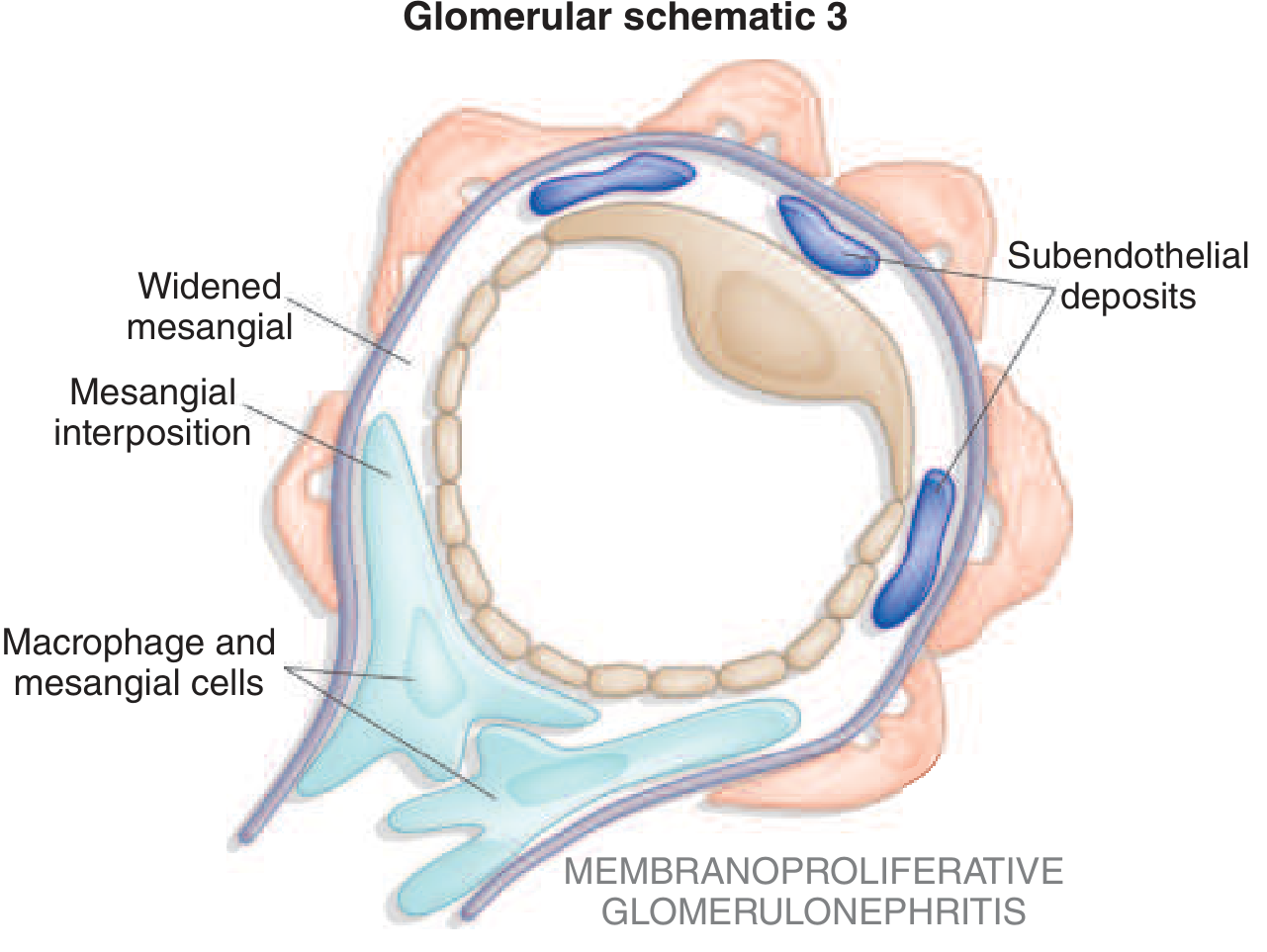
Key structural features: widened mesangium, mesangial interposition, subendothelial deposits, and macrophage/mesangial cell infiltration.
Pathology
Light Microscopy (the "MPGN pattern"):
- Lobular accentuation of glomerular tufts
- GBM thickening with double contours / tram-track sign (best seen on Jones' silver or PAS stain)
- Mesangial and endocapillary hypercellularity, infiltrating leukocytes
- Hyaline thrombi in capillary lumens suggest cryoglobulinemia or lupus
- Crescents may be present (>50% crescent formation = worse prognosis)
Immunofluorescence: Granular IgG + C3 deposits (IC-MPGN) or C3 dominant staining (C3G)
Electron Microscopy: Subendothelial and mesangial electron-dense deposits (Type I); intramembranous dense deposits replacing the GBM (DDD/Type II)
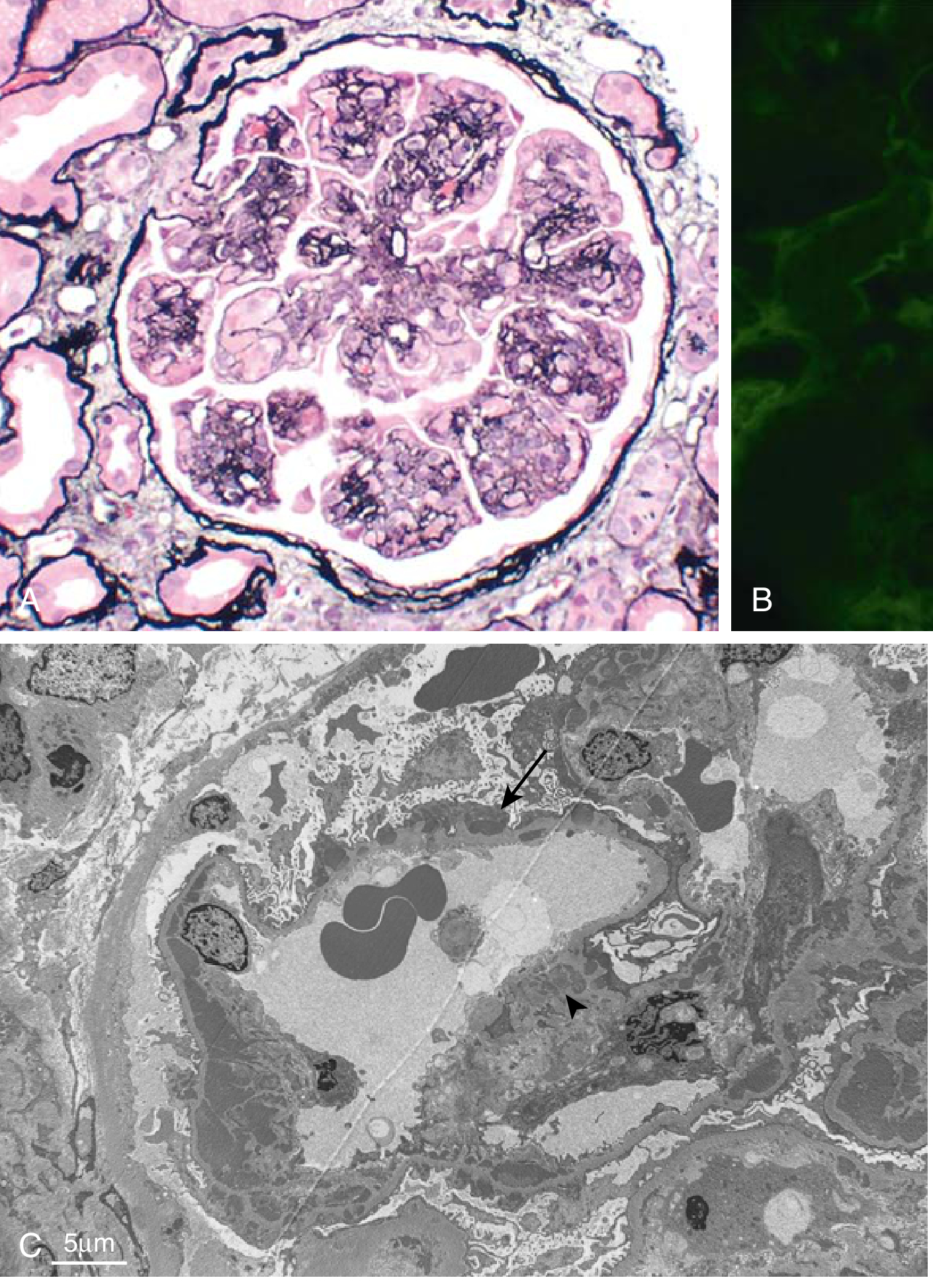
Fig A: Mesangial proliferation, GBM splitting (tram-tracking), lobular architecture. Fig B: Granular IgG deposits on IF. Fig C: EM with subendothelial deposits (arrow) and mesangial deposits (arrowhead).
- Robbins & Kumar Basic Pathology, p. 507
Causes (Etiology)
IC-MPGN - Secondary Causes:
| Category | Examples |
|---|---|
| Infections | HCV (most common), HBV, subacute bacterial endocarditis, shunt nephritis, malaria, schistosomiasis |
| Autoimmune / Rheumatologic | SLE, Sjögren syndrome, scleroderma, mixed cryoglobulinemia |
| Monoclonal gammopathy | Multiple myeloma, MGUS, lymphoma, leukemia |
| Malignancy | Carcinoma, lymphoma |
| Inherited | α1-antitrypsin deficiency, C2/C3 deficiency |
C3 Glomerulopathy - Mechanism:
Dysregulation of the alternative complement pathway leads to uncontrolled C3 activation via:
- C3 nephritic factor (C3NeF) - autoantibody that stabilizes C3 convertase (C3bBb), prolonging its half-life 10-fold - the most common acquired cause
- Factor H deficiency/mutation - Factor H is the key fluid-phase regulator of alternative pathway
- Inhibitory autoantibodies to Factor H or other complement proteins (Factor B, Factor I)
- Genetic mutations in complement regulatory genes (CFH, CFI, CD46, C3, CFB)
- Brenner and Rector's The Kidney, p. 1376
Serologic Findings
| Marker | IC-MPGN (Type I) | DDD (Type II) |
|---|---|---|
| C3 | ↓↓ | ↓↓↓ (severely low) |
| C4 | N or ↓↓ | Normal |
| Cryoglobulins | ++ | - |
| C3NeF | ± | ++ (majority) |
| ANA, anti-dsDNA | For lupus-associated | - |
| HCV serology | Screen all patients | - |
| SPEP/SFLC | For monoclonal | - |
Low C4 with low C3 = classical pathway activation (immune complex) | Normal C4 with very low C3 = alternative pathway (C3G)
Clinical Presentation
- Typically adolescents and young adults (though any age)
- Mixed nephritic-nephrotic picture: hematuria + proteinuria (often nephrotic range)
- Hypertension, edema, reduced GFR
- Hypocomplementemia (especially C3)
- May present as RPGN if crescents are extensive
- ~50% reach ESRD within 10 years without treatment of underlying cause
Diagnosis
- Kidney biopsy - essential; LM + IF + EM together determine the category
- Complement workup: C3, C4, factor H level, factor H antibody, C3NeF, factor I
- Infection screen: HCV, HBV, blood cultures, ANA, ANCA
- Monoclonal protein: SPEP, UPEP, serum free light chains, bone marrow biopsy if needed
- Genetic panel for complement gene mutations (especially in C3G)
Treatment
Treatment is etiology-directed - there is no one-size-fits-all approach:
| Cause | Treatment |
|---|---|
| HCV-associated IC-MPGN | Direct-acting antivirals (DAAs) - highly effective; resolves disease in most cases |
| SLE-associated | Immunosuppression (mycophenolate, steroids, cyclophosphamide per class) |
| Cryoglobulinemia | Treat underlying HCV; rituximab for severe disease |
| Monoclonal gammopathy | Clone-directed therapy (proteasome inhibitors, anti-CD20) |
| C3 Glomerulopathy (C3G) | Immunosuppression (MMF ± steroids); terminal complement inhibitors (eculizumab) in selected cases; complement factor replacement under study |
| Idiopathic IC-MPGN | Steroids ± immunosuppression; limited evidence |
| All patients | ACEi/ARB for proteinuria reduction; SGLT2i for nephroprotection |
For transplant recipients with recurrent MPGN: IC-MPGN recurs in 20-45% of cases with up to 50% graft loss; HCV eradication pre-transplant substantially reduces recurrence.
- Comprehensive Clinical Nephrology 7th Ed., p. 1474
- NKF Primer on Kidney Diseases 8e
C3 Glomerulopathy Subtypes
| Feature | DDD (Dense Deposit Disease) | C3 GN |
|---|---|---|
| EM | Intramembranous dense deposits replacing GBM ("sausage-like") | Mesangial/subendothelial deposits; NO intramembranous deposits |
| C3NeF | 80-90% positive | ~50% positive |
| Complement | Very low C3, normal C4 | Low-normal C3, normal C4 |
| Prognosis | ~50% ESRD in 10 years; high recurrence post-transplant | Variable |
| Partial lipodystrophy | Associated (15% of DDD) | Rare |
Prognosis
- IC-MPGN: Better with treatment of underlying cause (especially HCV cure with DAAs); ~50% reach ESRD in 10 years if idiopathic
- DDD: ~50% reach ESRD within 10 years; nearly universal recurrence after transplant
- C3 GN: Variable; worse outcomes with crescents, tubulointerstitial fibrosis on biopsy
- Poor prognostic factors: nephrotic-range proteinuria at presentation, reduced GFR, crescents, interstitial fibrosis
Recent Evidence (2024-2026)
- Yu et al. (2024) - Updated review of MPGN as a pattern of injury, emphasizing systematic etiologic workup [PMID 39004461]
- Sethi S. (2026) - Kidney International review on "Finding the etiology of MPGN" - reinforces that thorough etiologic workup (including proteomics and genetic complement testing) changes management in many previously "idiopathic" cases [PMID 41895686]
FSGS
Finding Sources
Searching PubMed
focal segmental glomerulosclerosis FSGS pathogenesis treatment
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Finding Sources
Reading File
Reading File
I have comprehensive content across all domains. Let me compile the full answer now.
Focal Segmental Glomerulosclerosis (FSGS)
FSGS is a histopathological pattern of podocyte injury - not a single disease - characterized by focal (affecting some glomeruli) and segmental (affecting a portion of each affected glomerulus) areas of glomerular scarring/consolidation. It is the most common cause of idiopathic nephrotic syndrome in adults of African descent and accounts for ~25% of adult nephrotic syndrome overall.
Epidemiology
-
~10% of glomerulonephritis in general; up to 25% of adult nephrotic syndrome
-
Most common in African Americans (Black Americans disproportionately affected due to APOL1 risk alleles)
-
Incidence has increased 13-fold over the past three decades (even among Caucasians)
-
Can occur in children (<15% of pediatric nephrotic syndrome) and adults
-
Brenner and Rector's The Kidney, p. 1347
-
NKF Primer on Kidney Diseases 8e, p. 190
Classification (Etiologic)
| Type | Mechanism |
|---|---|
| Primary (Idiopathic) | Circulating glomerular permeability factor(s) causing direct podocyte injury; as-yet unidentified |
| Secondary | Viral, drug, or adaptive (hyperfiltration) injury to podocytes |
| Genetic | Mutations in podocyte structural proteins |
Secondary FSGS causes:
| Category | Examples |
|---|---|
| Viral | HIV-1 (collapsing variant = HIVAN), SARS-CoV-2 (with APOL1 high-risk genotype), Parvovirus B19, CMV, EBV, HCV |
| Drug-induced | Heroin (adulterants), interferon (α/β/γ), pamidronate, sirolimus, calcineurin inhibitors, anthracyclines, anabolic steroids, NSAIDs, lithium, direct-acting antivirals (ledipasvir/sofosbuvir) |
| Adaptive / Hyperfiltration | Obesity-related glomerulopathy, unilateral renal agenesis, reflux nephropathy, partial nephrectomy, low birth weight/oligomeganephronia, sickle cell disease, hypertensive nephrosclerosis, aging kidney |
| Other | Healing phase of focal proliferative GN, thrombotic microangiopathy, body-building |
- Goldman-Cecil Medicine, p. 1253
Genetic Forms
Genetic FSGS is steroid-resistant and generally does not recur after transplant (unlike primary FSGS). Key genes:
| Gene | Protein | Inheritance |
|---|---|---|
| NPHS2 | Podocin | AR - most common genetic cause of steroid-resistant NS |
| NPHS1 | Nephrin | AR - Finnish congenital NS |
| ACTN4 | α-actinin-4 | AD |
| TRPC6 | TRP cation channel 6 | AD |
| INF2 | Inverted formin 2 | AD |
| WT1 | Wilms' tumor suppressor | AD, de novo |
| APOL1 | Apolipoprotein L1 | AR (biallelic G1/G2 risk alleles) - 10-20x increased risk in Black Americans; arose because heterozygosity protects against trypanosomiasis |
| COL4A3/COL4A4 | Type IV collagen | AR/AD - emerging as most common genetic FSGS overall |
| MYH9, CD2AP, PLCE1, COQ2/6, SCARB2, LAMB2, others | Various podocyte proteins | AR or AD |
- Brenner and Rector's The Kidney, p. 1349 (Table 31.4)
- Goldman-Cecil Medicine, p. 1253
Pathogenesis
Primary FSGS:
- A circulating podocyte permeability factor (still unidentified despite candidates including suPAR, B7-1/CD80, and cardiotrophin-like cytokine 1/CLC1) causes foot process effacement and proteinuria
- Evidence: plasma exchange or protein A adsorption column causes remission of recurrent FSGS post-transplant
- MCD and primary FSGS may represent a disease spectrum - FSGS can evolve from an initial MCD-like phase
Secondary FSGS (Adaptive):
- Reduced nephron mass → compensatory glomerular hypertrophy → glomerular hypertension → podocyte stress → segmental sclerosis (perihilar predilection)
- Absence of hypoalbuminemia and subnephrotic proteinuria suggest secondary/adaptive etiology
Genetic FSGS:
-
Mutations in podocyte structural components of the slit diaphragm, actin cytoskeleton, or GBM anchoring proteins
-
Comprehensive Clinical Nephrology 7th Ed., p. 272
Morphology (Columbia Classification - 5 Variants)
The Columbia classification defines five FSGS variants with prognostic significance:
| Variant | Light Microscopy Appearance | Clinical Association | Prognosis |
|---|---|---|---|
| Collapsing | Segmental/global capillary collapse; hypertrophied/hyperplastic podocytes with prominent resorption droplets; microcystic tubular change | HIV (HIVAN), SARS-CoV-2 + APOL1, IV drug use, interferon; 91% African American in one series | Worst - ESKD in 65%; remission only 13% |
| Tip Lesion | Consolidation at the tubular pole (proximal tubule origin); foam cells, endothelial swelling | More often Caucasian; typically primary; often presents with heavy nephrosis | Best - ESKD only 6%; remission 76% |
| Cellular | Endocapillary hypercellularity (foam cells, inflammatory cells) distributed throughout the tuft, not confined to tip or hilum | Primary FSGS | Intermediate - ESKD ~28%; remission 44% |
| Perihilar | Sclerosis and hyalinosis at the glomerular hilum | Secondary/adaptive FSGS (hyperfiltration); solitary kidney, obesity | Intermediate |
| NOS (Not Otherwise Specified) | Sclerosis not meeting other criteria; most common variant | Mixed primary and secondary | Intermediate - ESKD ~35%; remission 39% |
Immunofluorescence: IgM and C3 trapped (non-specifically) in areas of sclerosis; no immune deposits in primary/genetic FSGS
Electron microscopy:
-
Diffuse/extensive foot process effacement (>80% of capillary surface) = primary FSGS
-
Segmental/focal foot process effacement = secondary (adaptive) FSGS
-
No immune-type deposits in primary/genetic FSGS
-
Endothelial tubuloreticular inclusions (interferon signature) in >90% of HIV-associated collapsing FSGS
-
Brenner and Rector's The Kidney, p. 1347-1348
Clinical Features
| Feature | Primary FSGS | Secondary (Adaptive) FSGS |
|---|---|---|
| Proteinuria | Heavy, often nephrotic range | Subnephrotic to nephrotic |
| Hypoalbuminemia | Present | Often absent |
| Edema | Yes | Less pronounced |
| Hypertension | 30-50% | Common |
| Microscopic hematuria | ~50% | Less common |
| GFR at presentation | Reduced in 20-30% | Variable |
| Serum complement | Normal | Normal |
| Foot process effacement on EM | Diffuse (>80%) | Segmental |
Diagnosis
- Kidney biopsy is required - LM + IF + EM
- Serologic workup: complement (normal in FSGS), ANA, ANCA, anti-GBM (usually normal)
- HIV serology (mandatory)
- Assess for secondary causes: BMI/obesity, medications, nephron mass reduction history
- Genetic testing: consider in children, young adults, family history of kidney disease, steroid-resistant NS, atypical presentation - COL4A gene panel + podocyte gene panel
- suPAR level (elevated in some primary FSGS; not yet validated as clinical test)
Treatment
First determine: Primary vs. Secondary vs. Genetic
| Situation | Approach |
|---|---|
| Secondary/Adaptive FSGS | Treat underlying cause; ACEi/ARB; no immunosuppression |
| Genetic FSGS | Conservative management: ACEi/ARB; no immunosuppression (steroid-resistant) |
| Primary FSGS with nephrotic syndrome | Immunosuppression (below) |
| Subnephrotic proteinuria (any type) | ACEi/ARB; SGLT2i; no immunosuppression |
Primary FSGS - Treatment Ladder:
1st line: Corticosteroids
- Prednisone 1 mg/kg/day (max 80 mg/day) or 2 mg/kg every other day (max 120 mg)
- Minimum 3-4 months; taper over 6 months if remission
- Complete or partial remission in ~40-60% of patients
- Steroid-resistant = no improvement within 4 months
2nd line (steroid-resistant): Calcineurin Inhibitors (CNI)
- Cyclosporine 5-6 mg/kg/day OR tacrolimus 0.05-0.1 mg/kg/day
- Minimum 6 months before declaring CNI-resistance
- If remission: continue minimum 12 months then slow taper
- Monitor renal function and drug levels (nephrotoxicity risk)
Steroid-dependent/frequently relapsing:
- Rituximab (two 1g doses 1 week apart) - effective in steroid-dependent disease; less effective in steroid-resistant
- Mycophenolate mofetil (MMF) - steroid-sparing
All patients:
- ACEi or ARB - reduces proteinuria and slows progression
- SGLT2 inhibitors - nephroprotective
- Statins for hyperlipidemia
- Diuretics for edema (avoid volume depletion)
- Anticoagulation if thromboembolism risk high
HIVAN (HIV-associated collapsing FSGS):
-
Antiretroviral therapy (ART) is primary treatment - can partially reverse glomerular injury
-
ACEi/ARB adjunctive
-
Goldman-Cecil Medicine, p. 1253-1254
-
Comprehensive Clinical Nephrology 7th Ed., p. 272-273
Prognosis
| Factor | Worse Prognosis | Better Prognosis |
|---|---|---|
| Histologic variant | Collapsing (ESKD 65%) | Tip lesion (ESKD 6%) |
| Race | African American (faster progression) | - |
| Proteinuria | Nephrotic-range, persistent | Remission achieved |
| GFR at presentation | Reduced | Preserved |
| Histology | Interstitial fibrosis | Minimal fibrosis |
| Genetics | APOL1 high-risk, NPHS2 mutations | - |
- Untreated primary FSGS: ~50% reach ESRD within 10 years
- Sustained complete remission = excellent long-term kidney survival
- Partial remission also substantially reduces risk of ESRD
- Spontaneous remission is uncommon
Transplant:
-
Primary FSGS recurs in up to 30% of allografts (early, often within days)
-
Higher recurrence risk: younger patients, rapid progression to ESKD, prior allograft recurrence
-
Genetic FSGS: generally does NOT recur post-transplant
-
Goldman-Cecil Medicine, p. 1254
-
Comprehensive Clinical Nephrology 7th Ed., p. 272
Recent Evidence (2024-2026)
- Suresh et al. (2024) - Comprehensive FSGS review emphasizing the importance of differentiating primary/genetic/secondary before treatment [PMID 39084753]
- Altintas et al. (2025) - Annual Review of Pathology review on FSGS pathogenesis - updates on APOL1, circulating permeability factors, podocyte biology [PMID 39854184]
Approach to purpura
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.