all about absorption and distribution of drug
Reading File
Finding Sources
Searching the Web
drug absorption and distribution pharmacokinetics overview 2024
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have all the content and diagrams needed. Let me compile a comprehensive, well-structured answer.
Drug Absorption and Distribution - Complete Guide
Source: Lippincott Illustrated Reviews: Pharmacology (6th Ed.) & Katzung's Basic and Clinical Pharmacology (16th Ed.)
Overview: The ADME Framework
Pharmacokinetics describes what the body does to a drug. Every drug undergoes four sequential processes - Absorption, Distribution, Metabolism, and Excretion (ADME):
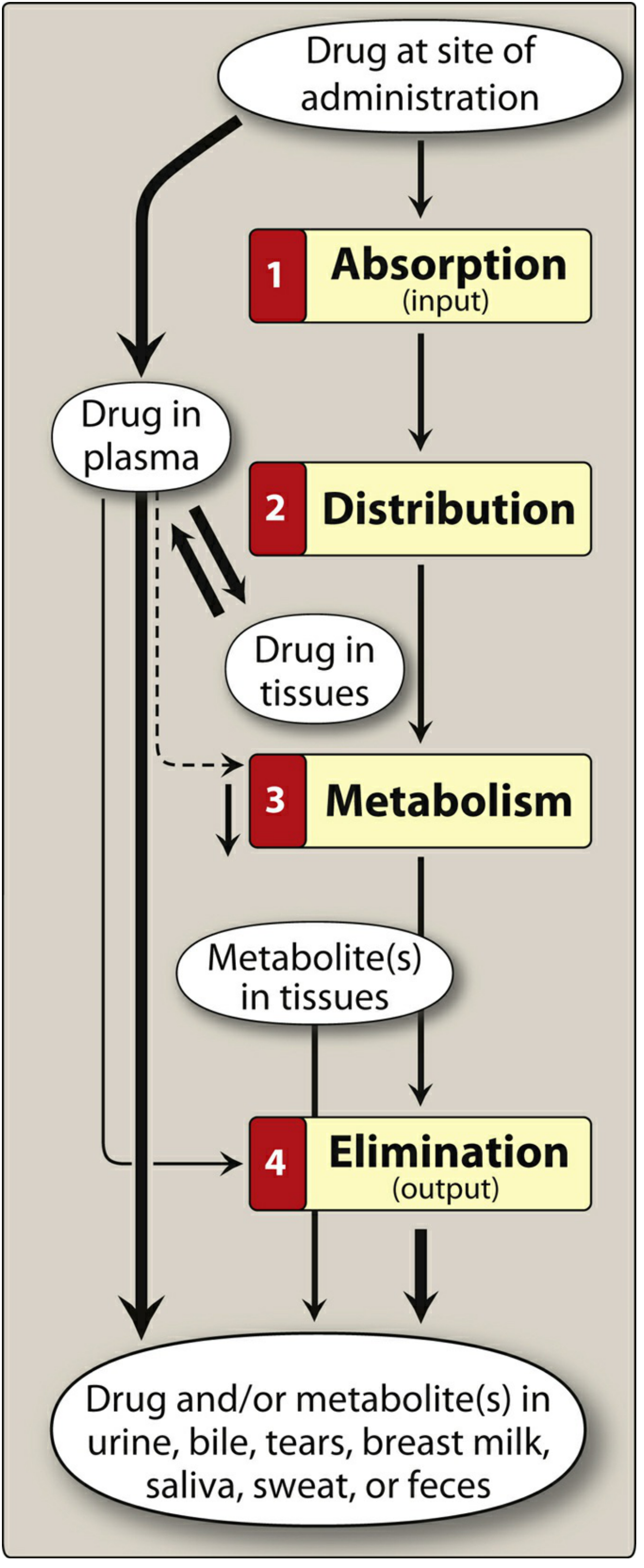
PART 1: DRUG ABSORPTION
Definition
Absorption is the transfer of a drug from its site of administration into the systemic (plasma) circulation. It is the first step determining when and how much drug reaches its target.
Routes of Administration
| Route | Absorption Pattern | Advantages | Disadvantages | Examples |
|---|---|---|---|---|
| Oral | Variable; many factors affect it | Convenient, economical | First-pass metabolism; food effects | Acetaminophen, amoxicillin |
| Sublingual | Rapid direct systemic absorption | Bypasses first-pass; avoids gastric pH | Limited to small doses | Nitroglycerin, buprenorphine |
| IV | Absorption not required (100% bioavailability) | Immediate effects; precise dosing | Infection risk; cannot be recalled | Morphine, heparin |
| IM | Relatively rapid | Suitable for poorly absorbed drugs | Painful; variable with blood flow | Diazepam, penicillin |
| SC | Slow, sustained | Good for poorly soluble drugs | Slow absorption | Insulin |
| Inhalation | Rapid (large surface area) | Direct delivery to lungs; minimal systemic SE | Requires proper technique | Albuterol, steroids |
| Transdermal | Slow; variable by skin site | Sustained release; avoids first-pass | Only for lipid-soluble drugs | Nicotine patch, fentanyl |
| Rectal | Erratic and incomplete | Useful if patient vomiting; 50% bypasses portal | Often irritating; erratic absorption | Diazepam rectal gel |
Key clinical point: IV administration gives 100% bioavailability by definition, and is the reference standard against which all other routes are compared.
Mechanisms of GI Absorption
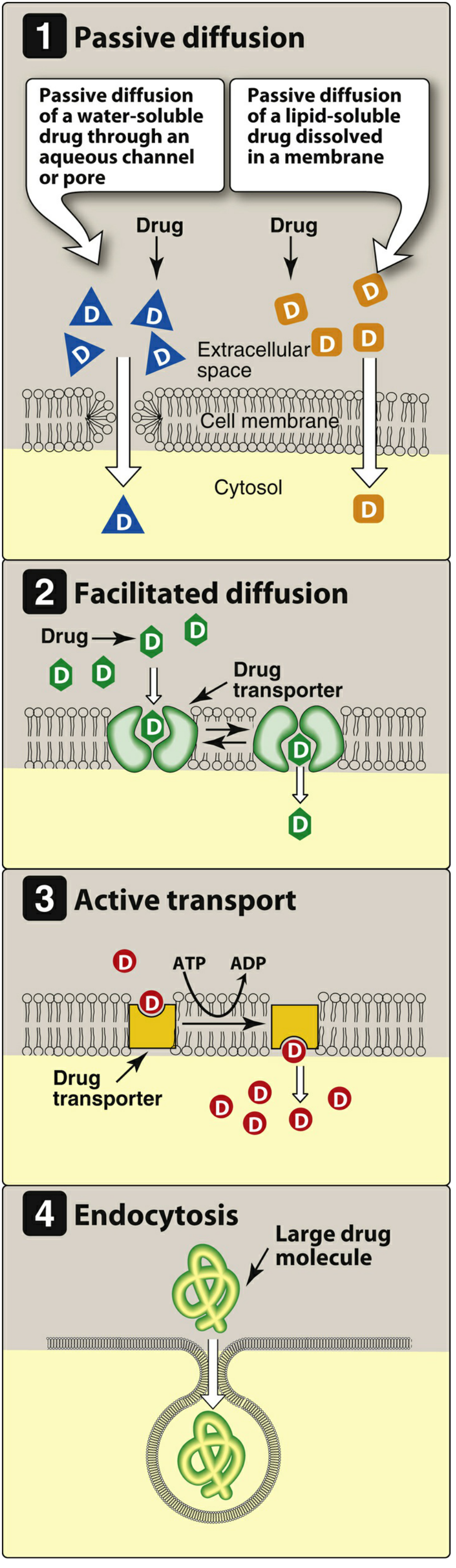
1. Passive Diffusion
- The most common mechanism for most drugs
- Driven by concentration gradient (high → low)
- No carrier protein needed; not saturable; low structural specificity
- Water-soluble drugs pass through aqueous channels/pores
- Lipid-soluble drugs dissolve directly through the lipid bilayer
2. Facilitated Diffusion
- Uses specialized transmembrane carrier proteins
- No energy required; moves drug down its concentration gradient
- Can be saturated and competitively inhibited
- Used for large molecules that cannot passively diffuse
3. Active Transport
- Carrier-protein mediated, energy-dependent (ATP hydrolysis)
- Can move drugs against a concentration gradient
- Saturable and selectively inhibited
- Example: levodopa is actively transported into the brain
4. Endocytosis
- Used for very large molecules (e.g., Vitamin B12)
- Cell membrane engulfs the drug, forms an intracellular vesicle
- Exocytosis is the reverse - used to secrete substances (e.g., norepinephrine from nerve terminals)
Factors Influencing Absorption
A. pH and Ionization (Henderson-Hasselbalch)
Drugs cross membranes far more readily in their uncharged (non-ionized) form.
- Weak acid (HA): HA ⇌ H⁺ + A⁻ → the uncharged form HA crosses membranes
- Weak base (BH⁺): BH⁺ ⇌ B + H⁺ → the uncharged form B crosses membranes
Practical consequences:
- Weak acids (aspirin, pKa ~3.5) are better absorbed in the acidic stomach where they remain mostly non-ionized
- Weak bases (morphine, pKa ~8) are better absorbed in the alkaline small intestine
- Ion trapping: A charged drug becomes "trapped" on the side of the membrane where it ionizes - e.g., basic drugs accumulate in acidic urine
B. Drug Solubility
- Very hydrophilic drugs: poor absorption due to inability to cross lipid membranes
- Very lipophilic drugs: also poor absorption due to insolubility in aqueous fluids at cell surfaces
- Ideal drugs: largely lipophilic but with some aqueous solubility - this is why many drugs are weak acids or weak bases
C. Chemical Instability
- Penicillin G: destroyed by gastric acid (low pH)
- Insulin: destroyed by GI proteolytic enzymes
- Such drugs require non-oral routes
D. Drug Formulation
Particle size, salt form, crystal polymorphism, enteric coatings, and excipients all affect dissolution rate and absorption speed.
Bioavailability
Bioavailability (F) is the rate and extent to which a drug reaches the systemic circulation unchanged.
- IV administration = 100% bioavailability by definition
- Measured by comparing the Area Under the Curve (AUC) of oral vs. IV administration
Formula:
F = AUC(oral) / AUC(IV) × 100%
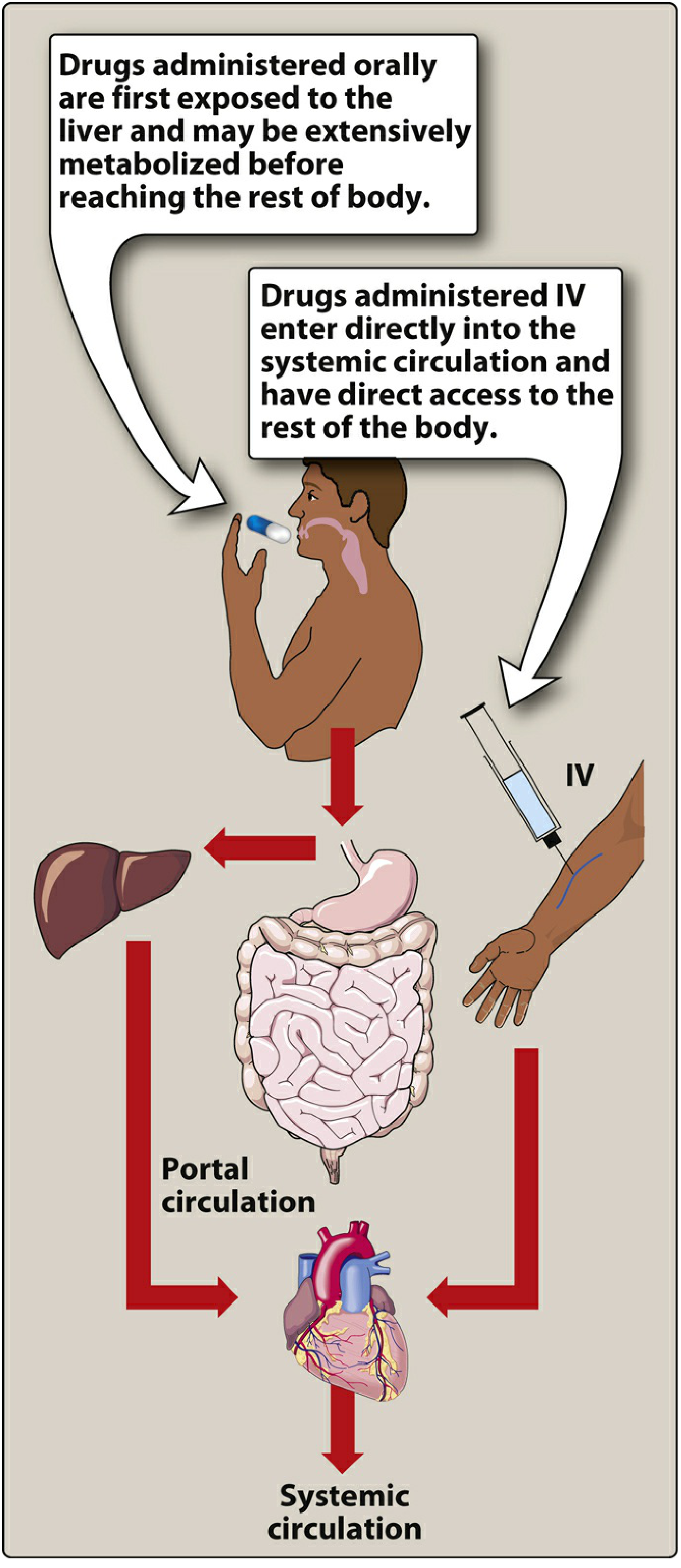
First-Pass Hepatic Metabolism
After oral absorption, drug enters the portal circulation before reaching systemic circulation. If the liver rapidly metabolizes it during this first pass, systemic bioavailability is dramatically reduced.
Classic examples:
- Nitroglycerin: >90% cleared on first pass → given sublingually, transdermally, or IV
- Lidocaine: extensive first-pass → not given orally
- Morphine: ~33% oral bioavailability due to first-pass
PART 2: DRUG DISTRIBUTION
Definition
Distribution is the reversible process by which a drug leaves the bloodstream and enters the extracellular fluid and tissues. It determines:
- Which tissues the drug reaches
- The concentration at the site of action
- Duration of drug effect
Key determinants of distribution:
- Cardiac output and local blood flow
- Capillary permeability
- Tissue volume
- Protein binding (plasma and tissue)
- Lipophilicity of the drug
A. Blood Flow
Blood flow varies dramatically by tissue:
| Tissue Type | Blood Flow | Example Drugs |
|---|---|---|
| Vessel-rich organs (brain, liver, kidney, heart) | Very high | Propofol - rapid CNS entry |
| Skeletal muscle | Moderate | Most IM-injected drugs |
| Adipose tissue, skin, viscera | Low | Highly lipophilic drugs accumulate here |
Clinical example: Propofol (highly lipophilic) rapidly distributes into the CNS (producing anesthesia) due to high blood flow + lipophilicity. Then slowly redistributes to fat and muscle, lowering plasma concentration, causing drug to exit the CNS → consciousness returns.
B. Capillary Permeability and the Blood-Brain Barrier (BBB)
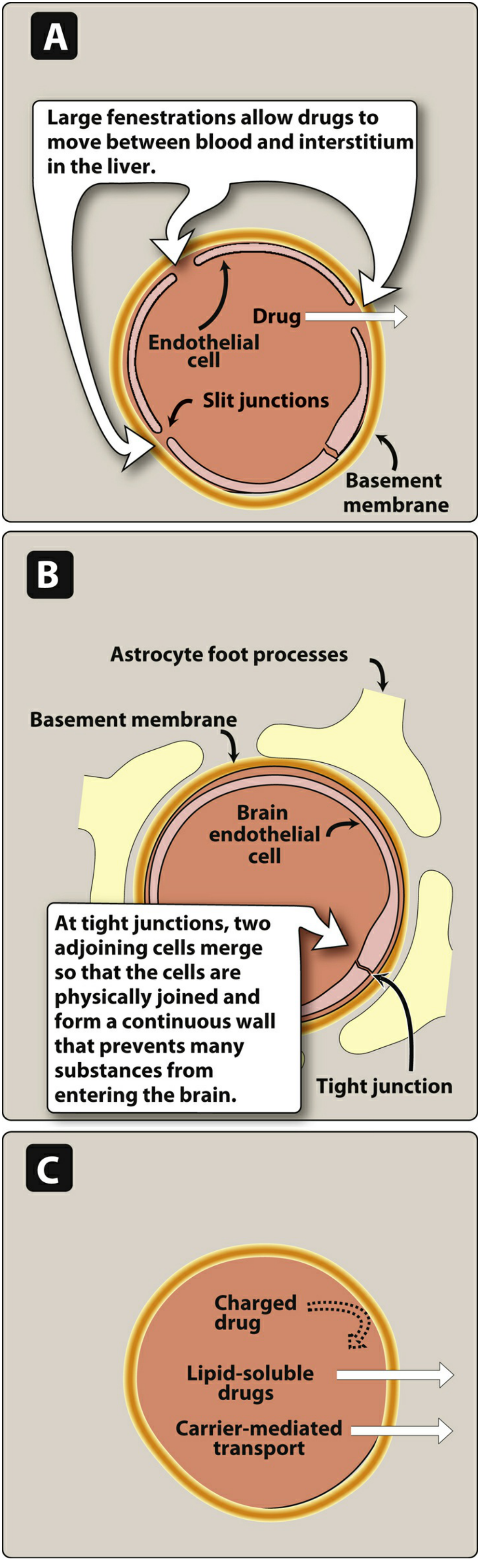
- Liver/spleen capillaries: Large fenestrations with slit junctions → even large plasma proteins can pass through
- Brain capillaries (BBB): Continuous endothelium with tight junctions and astrocyte foot processes → no slit junctions
What crosses the BBB:
- ✅ Lipid-soluble drugs (dissolve through endothelial cell membranes)
- ✅ Carrier-transported drugs (e.g., levodopa via large neutral amino acid transporter)
- ❌ Ionized/polar drugs
- ❌ Large-molecule drugs
Clinical significance: Antibiotics like penicillin penetrate the BBB poorly under normal conditions, but inflammation (meningitis) increases permeability and allows some entry.
C. Plasma Protein Binding
The major plasma binding proteins are:
- Albumin - binds most acidic drugs (warfarin, NSAIDs, penicillin) and some basic drugs
- Alpha-1-acid glycoprotein (AAG) - binds basic drugs (lidocaine, propranolol)
- Globulins - bind thyroid hormones, steroid hormones
Key Principles:
- Only free (unbound) drug is pharmacologically active - bound drug cannot reach receptors, be metabolized, or be excreted
- Protein binding acts as a drug reservoir - bound drug dissociates as free drug is eliminated
- Binding is reversible and typically in equilibrium
- Binding is saturable at high drug concentrations
Drug Displacement Interactions:
When two drugs compete for the same binding sites, one drug can displace the other. The displaced drug has increased free concentration → increased effect or toxicity.
Example: Warfarin (99% protein-bound) displaced by aspirin → sudden rise in free warfarin → bleeding risk.
Effect of Disease on Protein Binding:
| Condition | Effect | Mechanism |
|---|---|---|
| Hypoalbuminemia (cirrhosis, nephrotic syndrome) | Increased free drug | Less albumin available |
| Uremia (renal failure) | Decreased binding | Accumulated uremic acids compete with drugs |
| Inflammation/acute phase | Increased AAG | Basic drugs more bound |
D. Volume of Distribution (Vd)
Vd is an apparent volume that relates the total amount of drug in the body to the plasma concentration:
Vd = Total amount of drug in body / Plasma concentration
It is a mathematical concept, not a real anatomical volume. It tells you how extensively a drug distributes into tissues.
The Three Body Water Compartments:
| Compartment | Volume (70-kg adult) | Drug type | Examples |
|---|---|---|---|
| Plasma | ~4 L | High MW or extensively protein-bound | Heparin (Vd ~4 L) |
| Extracellular fluid (plasma + interstitium) | ~14 L | Low MW but hydrophilic | Aminoglycosides (Vd ~14 L) |
| Total body water | ~42 L | Low MW and lipophilic | Ethanol (Vd ~42 L) |
| Beyond total body water | >42 L | Sequestered in tissues/fat | Chloroquine (Vd >200 L!), digoxin |
Interpreting Vd:
- Small Vd (3-5 L): Drug stays in plasma - likely large or highly protein-bound (e.g., heparin, warfarin)
- Moderate Vd (10-20 L): Distributes into extracellular fluid (e.g., gentamicin)
- Large Vd (>42 L): Extensively tissue-distributed - may be hard to remove by dialysis (e.g., digoxin, tricyclic antidepressants)
Clinical Importance of Vd:
A large Vd increases the half-life of a drug - drug sequestered in tissues is unavailable to the liver/kidneys for elimination. This explains why drugs like amiodarone (Vd ~5000 L) take weeks to reach steady state and weeks to eliminate.
Loading dose formula:
Loading dose = Vd × Target plasma concentration
E. Tissue Binding and Sequestration
Some drugs accumulate in specific tissues:
| Drug | Site of Accumulation | Clinical Note |
|---|---|---|
| Chloroquine, amiodarone | Lung, liver, fat | Very large Vd; very long t½ |
| Tetracyclines | Bone, teeth | Avoided in children/pregnancy |
| Iodine | Thyroid | Used in thyroid scanning |
| Carbon monoxide | Hemoglobin | Toxic sequestration |
| DDT, thiopental | Adipose tissue | Prolonged effect in obese |
F. Special Populations and Distribution
Neonates:
- Higher total body water (70-85% vs. 50-60% in adults) → higher Vd for water-soluble drugs (e.g., aminoglycosides need higher mg/kg dose)
- Very little body fat (~1-15% vs. 20%+ in adults) → less accumulation of lipophilic drugs
- Reduced plasma protein binding → higher free drug concentrations at the same total plasma level → increased risk of toxicity
- BBB is more permeable → drugs like bilirubin and morphine reach CNS more readily
Elderly:
- Decreased plasma albumin → higher free fractions of acidic drugs
- Decreased body water → higher concentration of water-soluble drugs
- Increased fat → larger Vd for lipophilic drugs → prolonged effect
Hepatic failure:
- Decreased albumin synthesis → more free drug
- Decreased first-pass metabolism → higher bioavailability of oral drugs
Summary: Absorption vs. Distribution
| Feature | Absorption | Distribution |
|---|---|---|
| Definition | Drug moving from site of administration into plasma | Drug moving from plasma into tissues |
| Direction | Into the blood | Out of the blood |
| Key determinant | Route of administration, pH, lipophilicity, first-pass | Blood flow, protein binding, Vd, BBB |
| Quantified by | Bioavailability (F) | Volume of distribution (Vd) |
| Clinically modified by | Route change (SL instead of oral), formulation | Protein binding displacement, disease states |
Quick-Reference: Key Drug Examples
| Drug | Absorption Quirk | Distribution Quirk |
|---|---|---|
| Nitroglycerin | >90% first-pass → sublingual only | Vd ~3 L (stays in plasma) |
| Warfarin | Well absorbed orally | 99% albumin-bound; small Vd |
| Gentamicin | Not absorbed orally (polar) | Distributes extracellular water; Vd ~14 L |
| Digoxin | Well absorbed orally | Very large Vd (~500 L); tissue-bound |
| Propofol | IV only | Highly lipophilic; rapid CNS entry |
| Chloroquine | Oral, good absorption | Vd >200 L; accumulates in tissues |
| Heparin | Not absorbed orally (large, charged) | Stays in plasma; Vd ~4 L |
Sources: Lippincott Illustrated Reviews: Pharmacology, Chapter 1 (Pharmacokinetics) | Katzung's Basic and Clinical Pharmacology, 16th Ed., Chapter 59 (Drug Therapy in Infants & Children)
This is a shared conversation. Sign in to Orris to start your own chat.