In fructose intolerance, ingestion of fructose leads to hypoglycemia and liver damage. Outline the metabolic pathway involved and the mechanism by which aldose B deficiency causes these symptoms.
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the information needed for a thorough, well-sourced answer. Here it is:
Hereditary Fructose Intolerance (HFI): Metabolic Pathway and Mechanism
Normal Fructose Metabolism
Fructose absorbed in the small intestine travels to the liver, which processes the vast majority of dietary fructose. The pathway proceeds as follows:
Step 1 - Phosphorylation by fructokinase:
Fructose + ATP → Fructose-1-phosphate (F1P) + ADP
Fructokinase has a high Vmax and low Km, meaning it rapidly and voraciously phosphorylates fructose as it enters hepatocytes. Critically, this step is unregulated - there is no feedback inhibition analogous to glucose's regulation by hexokinase or phosphofructokinase-1.
Step 2 - Cleavage by Aldolase B (the key enzyme):
Fructose-1-phosphate → Dihydroxyacetone phosphate (DHAP) + Glyceraldehyde
Aldolase B (found in liver, kidney, and small intestinal mucosa) is the only isoform capable of cleaving fructose-1-phosphate. Aldolase A (muscle) and aldolase C (brain) can only cleave fructose-1,6-bisphosphate (the glycolytic intermediate).
Step 3 - Entry into glycolysis:
- Glyceraldehyde is phosphorylated by triose kinase → Glyceraldehyde-3-phosphate
- DHAP and glyceraldehyde-3-phosphate are both glycolytic intermediates
These two triose phosphates then proceed through glycolysis to pyruvate, enter the TCA cycle, or are used for gluconeogenesis and fatty acid synthesis.
The pathway diagram from Basic Medical Biochemistry:
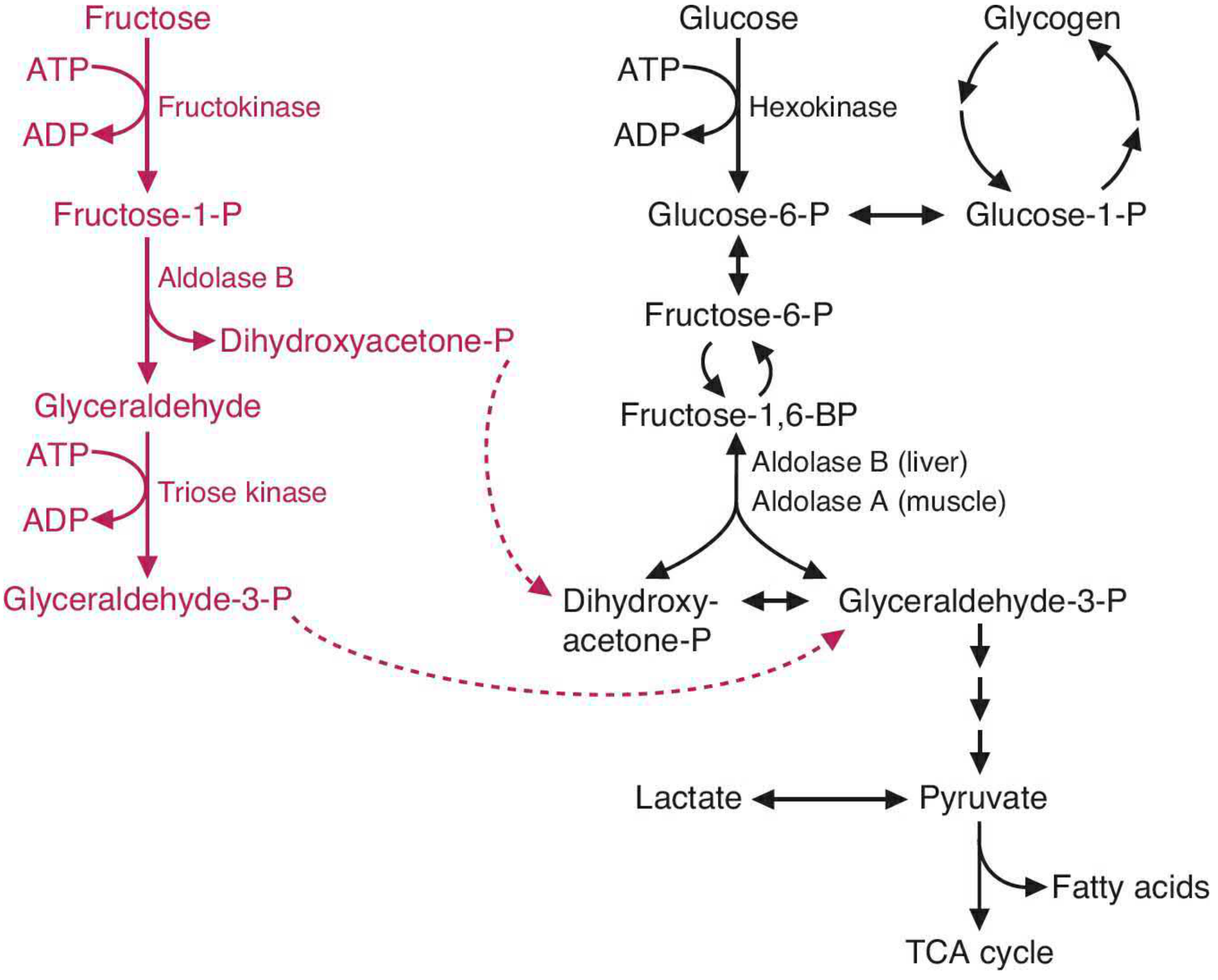
The Defect in HFI: Aldolase B Deficiency
HFI is an autosomal recessive disorder (incidence ~1:20,000 live births). The most common mutation in people of European descent is a missense mutation in exon 5 (G → C), causing an Ala → Pro substitution that produces catalytically impaired aldolase B. When fructose is ingested, the pathway stalls at fructose-1-phosphate, which accumulates massively in liver, kidney, and intestinal cells.
Mechanism of Hypoglycemia
The accumulation of F1P causes hypoglycemia through two convergent mechanisms:
1. Phosphate (Pi) sequestration and ATP depletion
Fructokinase continues consuming ATP and Pi to make more F1P, but aldolase B cannot clear it. This traps inorganic phosphate inside the F1P molecule, rapidly depleting free cytoplasmic Pi. With phosphate unavailable:
- Oxidative phosphorylation (which requires ADP + Pi → ATP) is severely impaired
- ATP falls sharply; AMP rises
2. Dual block on glucose production
With ATP depleted, the liver cannot maintain blood glucose through its two normal mechanisms:
- Gluconeogenesis is inhibited: ATP is required to drive gluconeogenesis (e.g., phosphorylation steps). The fall in hepatic ATP blocks new glucose synthesis.
- Glycogenolysis is inhibited: F1P (and to a lesser extent fructose-1,6-bisphosphate) allosterically inhibits liver glycogen phosphorylase, the enzyme that mobilizes glycogen to release glucose-1-phosphate. This is why hypoglycemia occurs despite normal or elevated glycogen stores - the glycogen simply cannot be released.
The result is a rapid, profound drop in blood glucose after fructose ingestion, presenting as pallor, sweating, vomiting, and in severe cases, convulsions, lethargy, and coma.
Mechanism of Liver (and Kidney) Damage
ATP depletion is the central driver of hepatocellular injury:
- Decreased protein synthesis: ATP is required for protein synthesis. Falling ATP leads to reduced synthesis of clotting factors (prolonged prothrombin time), albumin (hypoalbuminemia), and other essential hepatic proteins.
- Cell death: Sustained ATP depletion leads to cellular energy failure - failure of Na+/K+-ATPase pumps, cellular swelling, and ultimately necrosis. Over time this causes ultrastructural changes in the liver, progressing to hepatomegaly, jaundice (elevated bilirubin and transaminases), hepatic fibrosis, and eventually liver failure if fructose ingestion continues.
- Fanconi-like renal tubular dysfunction: Proximal tubule cells also have fructokinase and aldolase B. The same ATP-depletion mechanism impairs tubular reabsorption of Pi, glucose, amino acids, and urate - producing a Fanconi-like syndrome with phosphaturia, aminoaciduria, and nephrocalcinosis.
The AMP degradation cascade also contributes: as AMP rises from ATP depletion, it is catabolized to uric acid, producing hyperuricemia and contributing to lactic acidemia (as pyruvate is shunted to lactate when mitochondrial oxidative phosphorylation falters).
Why Essential Fructosuria is Benign by Comparison
A deficiency of fructokinase (essential fructosuria) is entirely benign because F1P never accumulates - fructose simply cannot be phosphorylated and is excreted in the urine. There is no Pi sequestration, no ATP depletion, and no downstream consequences. This contrast illustrates that the toxicity in HFI is due specifically to the accumulation of F1P, not to the absence of fructose catabolism per se.
Summary Table
| Feature | Mechanism |
|---|---|
| Hypoglycemia | Pi sequestration → ATP depletion → blocked gluconeogenesis; F1P allosterically inhibits glycogen phosphorylase |
| Liver damage | ATP depletion → reduced protein synthesis, Na+/K+ pump failure, cell necrosis; fibrosis with chronic exposure |
| Hyperuricemia | AMP degradation following ATP depletion |
| Lactic acidosis | Impaired oxidative phosphorylation shifts pyruvate to lactate |
| Renal tubular dysfunction | Same F1P/ATP mechanism in proximal tubule cells |
Treatment: Lifelong elimination of fructose, sucrose, and sorbitol (sorbitol is converted to fructose) from the diet. With early treatment, liver and kidney dysfunction are reversible and prognosis is good.
Sources: Lippincott Illustrated Reviews: Biochemistry, 8e, pp. 406-408; Basic Medical Biochemistry: A Clinical Approach, 6e, pp. 798-817; Harper's Illustrated Biochemistry, 32e, p. 211; Harrison's Principles of Internal Medicine, 22e, p. 3410
A child with recurrent hypoglycemia and seizures is diagnosed with glycogen storage disease type 3, Cori's disease. Discuss the enzymatic defect in rebranching enzyme and its impact on glycogen breakdown.
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Glycogen Storage Disease Type III (Cori's Disease): Enzymatic Defect and Mechanism
Background
Cori's disease (also called Forbes disease or limit dextrinosis) is an autosomal recessive disorder caused by mutations in the AGL gene, which encodes the glycogen debranching enzyme. It affects approximately 1:100,000 births and typically presents in infancy when the child begins fasting overnight - manifesting as the recurrent hypoglycemia and seizures described in this case. Both liver and muscle are usually affected (type IIIA, ~85% of cases); ~15% have liver involvement only (type IIIB).
Normal Glycogen Breakdown: The Role of the Debranching Enzyme
To understand the defect, the normal pathway of glycogenolysis must be clear:
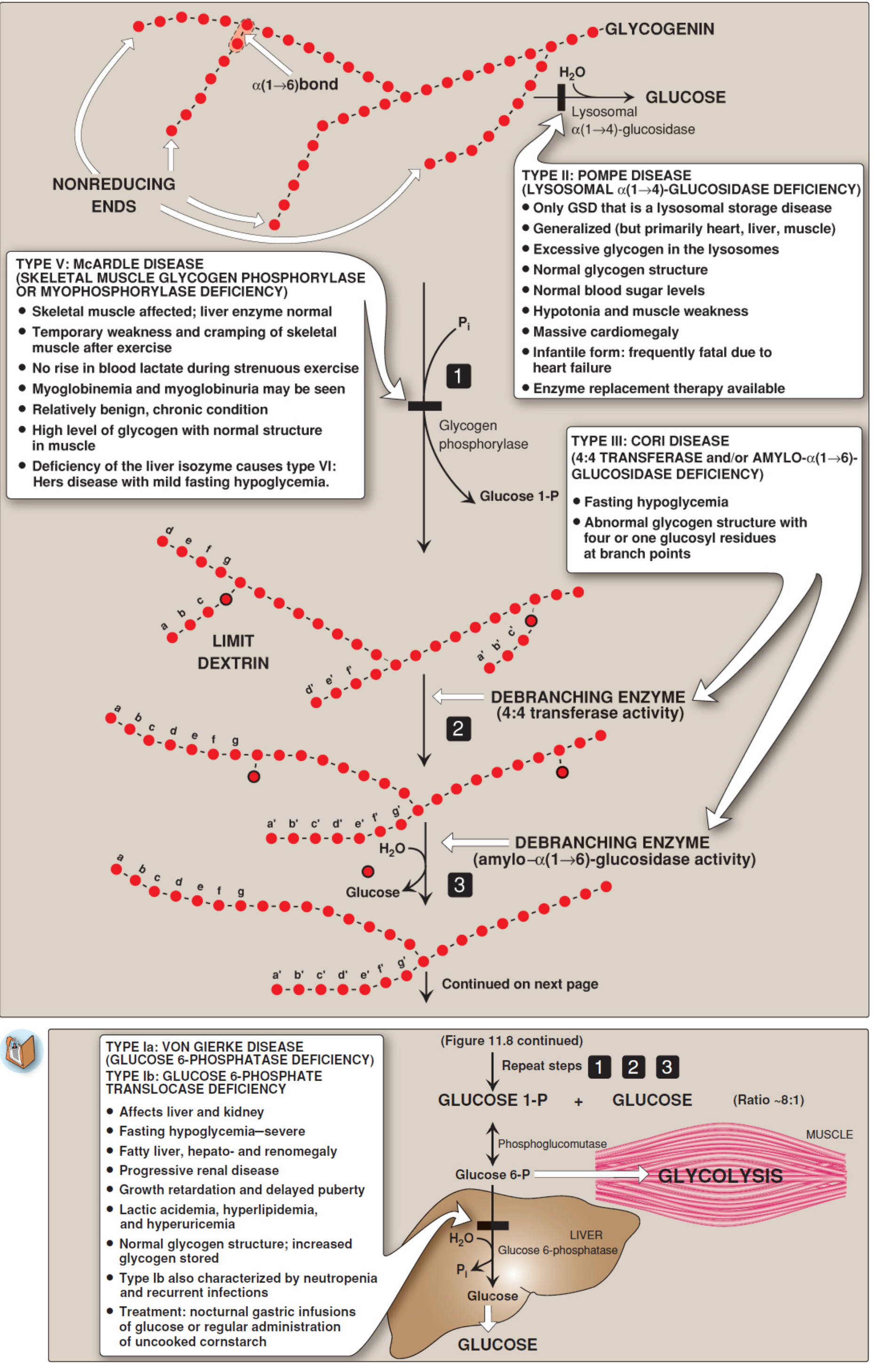
Step 1 - Glycogen phosphorylase cleaves α(1→4) glycosidic bonds at the nonreducing ends of glycogen chains by phosphorolysis, releasing glucose-1-phosphate. It proceeds sequentially inward until it reaches a point 4 glucosyl residues away from an α(1→6) branch point. At that point, phosphorylase stops completely. The resulting stupped structure is called a limit dextrin.
Step 2 - Debranching enzyme (4:4 transferase activity): The debranching enzyme is a single bifunctional polypeptide that carries out two distinct catalytic activities. First, its oligo-α(1→4)→α(1→4)-glucantransferase (4:4 transferase) activity removes the outer three of the four remaining glucosyl residues at the branch stub and transfers the trisaccharide unit to the free nonreducing end of another chain, extending it via a new α(1→4) bond. This leaves a single glucosyl residue attached at the branch point via an α(1→6) linkage.
Step 3 - Debranching enzyme (amylo-α(1→6)-glucosidase activity): The same enzyme's second active site then hydrolytically cleaves that lone α(1→6)-linked glucose residue, releasing it as free (unphosphorylated) glucose. The branch point is now resolved and the main chain is again a suitable substrate for phosphorylase.
Steps 1-3 repeat in cycles until glycogen is completely broken down. The ratio of glucose-1-phosphate to free glucose produced is approximately 8:1, reflecting that ~90% of glycosidic bonds are α(1→4) and ~10% are α(1→6).
The glucose-1-phosphate produced is isomerized by phosphoglucomutase to glucose-6-phosphate, which in the liver is dephosphorylated by glucose-6-phosphatase and released as free glucose into the blood. Muscle lacks glucose-6-phosphatase, so its glucose-6-phosphate enters local glycolysis instead.
The Enzymatic Defect in Cori's Disease
The debranching enzyme (gene: AGL) is deficient or dysfunctional. Either or both active sites may be affected:
| Subtype | Defect |
|---|---|
| IIIa | Both transferase and glucosidase activities absent (liver + muscle) |
| IIIb | Both activities absent in liver only |
| IIIc | Isolated glucosidase deficiency |
| IIId | Isolated transferase deficiency |
The most common form (IIIa) abolishes both activities in liver and muscle.
Consequences: How the Defect Causes Disease
When debranching enzyme is absent or non-functional:
- Phosphorylase proceeds normally down each outer chain until it reaches 4 residues from a branch point - then halts.
- The 4:4 transferase step cannot occur - the trisaccharide cannot be relocated to another chain.
- The α(1→6)-glucosidase step cannot occur - the branch point glucose cannot be freed.
- Glycogenolysis is therefore permanently arrested at every branch point. The liver and muscle accumulate a structurally abnormal form of glycogen - "limit dextrin" - characterized by short outer chains with 4 (or 1) glucosyl residues remaining at every branch point rather than the normal ~8-12 residue outer chains.
Mechanism of Hypoglycemia
The hypoglycemia in Cori's disease arises because the liver cannot fully mobilize glycogen stores during fasting:
- Glycogen phosphorylase can still act on the peripheral chains and initially provides some glucose-1-phosphate. However, once phosphorylase reaches the first tier of branch points and stalls, the deeper, inner core of glycogen is completely inaccessible.
- Because every branch in the glycogen molecule is a dead end, the vast majority of stored glucose cannot be released, even though liver glycogen content is actually elevated (the abnormal limit dextrin accumulates).
- Gluconeogenesis is intact (unlike GSD type I), so hypoglycemia is typically milder and more fasting-dependent than in Von Gierke disease. However, during prolonged fasting - as seen overnight in young children - blood glucose falls and seizures can result.
A key differentiating point from GSD type I: because gluconeogenesis is functional, a hyperglycemic response to galactose or glucagon administration can be demonstrated (galactose is converted to glucose via a gluconeogenic route). This response is absent in type I.
Mechanism of Hepatomegaly and Liver Damage
- Massive accumulation of limit dextrin (abnormal short-chain glycogen) causes hepatomegaly.
- Progressive hepatic glycogen storage leads to elevated transaminases (AST, ALT), and over time to hepatic fibrosis and cirrhosis in some patients.
- Hyperlipidemia is common as the liver shifts to fat mobilization during hypoglycemic episodes.
Mechanism of Muscle Disease (Type IIIA)
Muscle also expresses debranching enzyme and accumulates limit dextrin:
- The muscle cannot fully utilize glycogen for energy during exercise or fasting.
- This causes progressive proximal muscle weakness (which worsens with age, becoming more prominent in adults than children).
- Elevated creatine kinase (CK) reflects ongoing muscle damage.
- Unlike McArdle disease (GSD V), patients with Cori's disease do not typically develop acute exercise-induced cramps because some glycogen degradation still occurs (the outer chains can be phosphorylated by phosphorylase before it stalls). However, sustained energy demand during prolonged fasting can cause myopathy.
Summary of Clinical-Biochemical Correlations
| Feature | Mechanism |
|---|---|
| Fasting hypoglycemia | Glycogen phosphorylase halts at branch points; deeper glycogen inaccessible |
| Seizures | Cerebral neuroglycopenia from fasting hypoglycemia |
| Hepatomegaly | Accumulation of limit dextrin in hepatocytes |
| Elevated transaminases | Hepatocellular injury from glycogen overload |
| Hyperlipidemia | Compensatory fat mobilization during hypoglycemia |
| Muscle weakness (IIIA) | Limit dextrin accumulation in skeletal and cardiac muscle |
| Elevated CK | Myocellular injury |
| Normal lactate/urate | Gluconeogenesis intact; no glycolytic block (distinguishes from GSD I) |
| Abnormal glycogen structure | Short outer chains, normal branch frequency (limit dextrin) |
Comparison with GSD Type I (Von Gierke) - Key Distinction
In type I, glucose-6-phosphatase is absent, so glucose cannot be released even if glycogen is broken down normally. In type III, the defect is upstream: glycogen breakdown itself is blocked. This means:
- Gluconeogenesis is intact in type III (lactic acid and uric acid are not elevated)
- Galactose and fructose can raise blood glucose in type III (they bypass glycogen entirely)
- Hypoglycemia in type III is generally less severe than in type I
Treatment
- Frequent high-carbohydrate meals to prevent fasting hypoglycemia; uncooked cornstarch is used in older children for sustained glucose release
- High-protein diet in patients with significant myopathy (protein can fuel gluconeogenesis, bypassing the glycogen block entirely)
- Avoidance of prolonged fasting
- Liver transplantation has been performed for severe hepatic disease but does not reverse the muscle component
Sources: Lippincott Illustrated Reviews: Biochemistry, 8e, pp. 382-386; Basic Medical Biochemistry: A Clinical Approach, 6e, pp. 956-957; Harper's Illustrated Biochemistry, 32e, pp. 1509-1512; Tietz Textbook of Laboratory Medicine, 7e, p. 1145; Harrison's Principles of Internal Medicine, 22e, p. 3688
This is a shared conversation. Sign in to Orris to start your own chat.