Hey orris i want to study acute kidney injury from very basic to clinical also tell the symptoms which confirms the disease what kind of investigation should be asked and why explain me this in easy way and simplified version in short not in too much detail
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
I now have all the information I need. Here is a complete, simplified guide to AKI from basics to clinical.
Acute Kidney Injury (AKI) - From Basic to Clinical
1. What is AKI?
AKI is a rapid decline in kidney function that happens over hours to days. The kidney suddenly cannot filter waste from the blood properly, so toxic substances (like urea and creatinine) build up.
It was previously called "acute renal failure," but AKI is now the preferred term because the damage ranges from mild to severe and does not always mean complete failure.
KDIGO Definition (the gold-standard criteria):
You diagnose AKI when any one of these happens:
- Serum creatinine rises by ≥0.3 mg/dL within 48 hours, OR
- Serum creatinine rises ≥1.5× the baseline within 7 days, OR
- Urine output drops to <0.5 mL/kg/hr for ≥6 hours
2. The 3 Big Categories (Where Does the Problem Sit?)
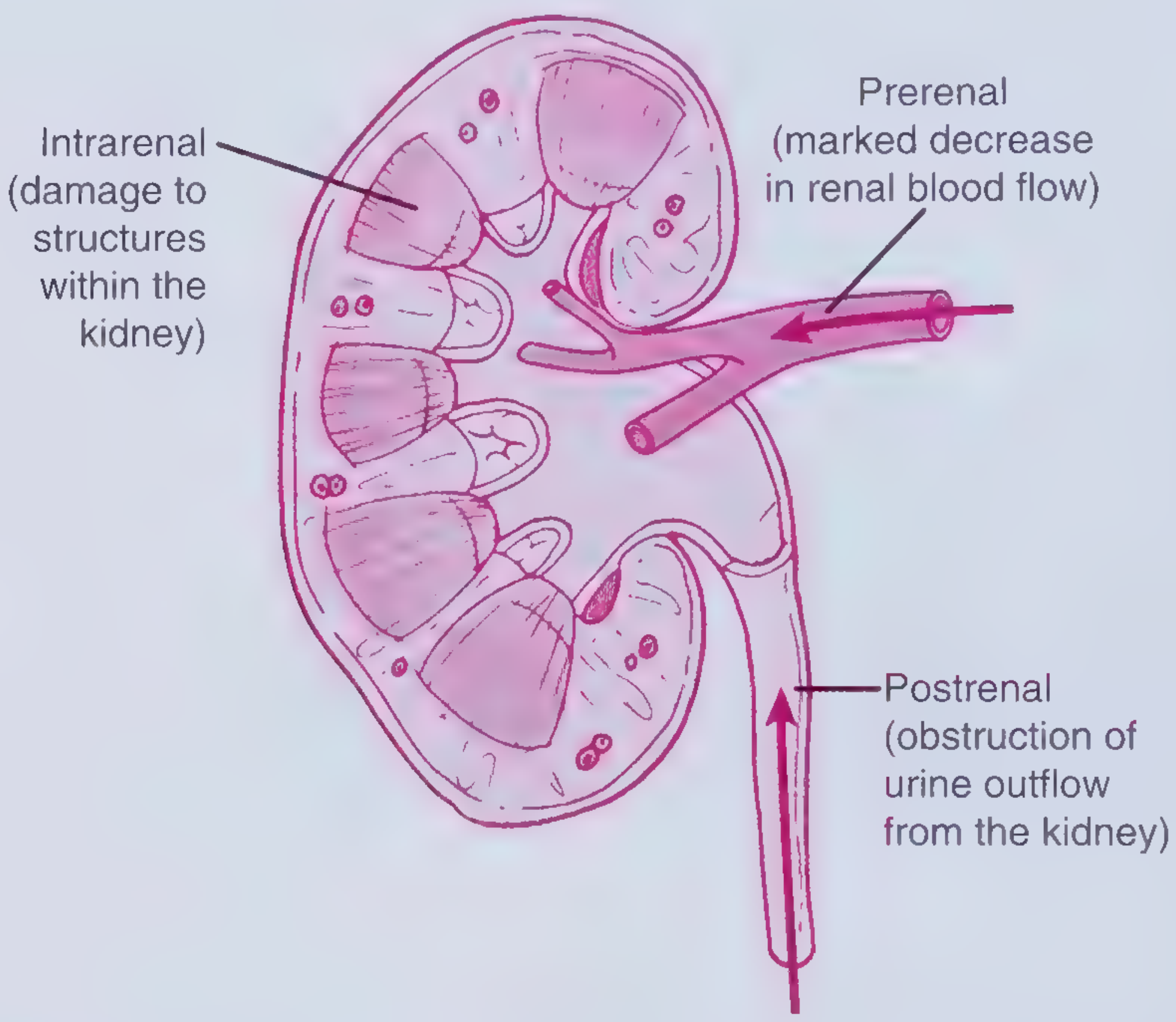
Think of the kidney like a factory. The problem can be:
- Before the factory (not enough blood coming in) = Prerenal
- Inside the factory (kidney tissue itself is damaged) = Intrinsic/Intrarenal
- After the factory (blockage to urine going out) = Postrenal

A. Prerenal AKI (most common, ~55-60%)
Mechanism: Not enough blood reaching the kidney. The kidney itself is not damaged yet - it is just "thirsty."
Common causes:
- Dehydration (vomiting, diarrhea, poor intake, excessive sweating)
- Blood loss (hemorrhage)
- Heart failure (pump is weak, low cardiac output)
- Sepsis (vasodilation - blood pressure drops)
- Liver failure / hepatorenal syndrome
- Diuretic overuse
- NSAIDs (constrict renal afferent arteriole)
- ACE inhibitors/ARBs (especially in bilateral renal artery stenosis)
Key point: If you restore blood flow quickly, the kidney recovers fully. If you ignore it, it progresses to actual kidney damage (ATN).
B. Intrinsic (Intrarenal) AKI (~35%)
The kidney tissue itself is damaged. This is subdivided by what part is hit:
| Part Damaged | Condition | Classic Cause |
|---|---|---|
| Tubules | Acute Tubular Necrosis (ATN) - most common | Prolonged ischemia (from unresolved prerenal), nephrotoxins (aminoglycosides, contrast, myoglobin from rhabdomyolysis) |
| Glomeruli | Glomerulonephritis | Immune diseases (IgA nephropathy, lupus, GPA/Wegener's) |
| Interstitium | Acute Interstitial Nephritis (AIN) | Drug allergy (penicillins, NSAIDs, PPIs), infections |
| Vessels | Vascular injury | TTP/HUS, malignant hypertension, atheroemboli |
ATN is the most important - it is the final common pathway when prerenal AKI is not treated in time. The tubule cells die from lack of oxygen (ischemic ATN) or from direct poison (toxic ATN).
C. Postrenal AKI (~5-10%)
Urine is made but cannot get out. Pressure backs up and damages both kidneys.
Common causes:
- Benign prostatic hyperplasia (BPH) - most common in older men
- Kidney stones (must block both ureters or a solitary kidney)
- Bladder or cervical cancer
- Strictures
- Neurogenic bladder
3. Staging of AKI (KDIGO)
| Stage | Serum Creatinine | Urine Output |
|---|---|---|
| 1 | 1.5-1.9× baseline OR rise ≥0.3 mg/dL | <0.5 mL/kg/hr for 6-12 hrs |
| 2 | 2.0-2.9× baseline | <0.5 mL/kg/hr for ≥12 hrs |
| 3 (most severe) | ≥3× baseline OR ≥4.0 mg/dL OR started on dialysis | <0.3 mL/kg/hr for ≥24 hrs OR anuria ≥12 hrs |
4. Symptoms - What the Patient Tells You
Symptoms arise from two things: fluid/waste retention and kidney not making urine.
Urinary symptoms:
- Oliguria - reduced urine (<400 mL/day) - the classic hallmark
- Anuria - no urine at all (very severe, or suggests postrenal obstruction)
- Frothy urine (if proteinuria present)
- Haematuria (blood in urine - suggests glomerulonephritis or stones)
Fluid overload symptoms (body retaining water):
- Swelling of legs, face (edema)
- Shortness of breath (pulmonary oedema - fluid in lungs)
- High blood pressure
Uraemic symptoms (waste products building up):
- Nausea and vomiting
- Confusion, drowsiness, lethargy
- Loss of appetite
- Muscle cramps
- Hiccups
- Itching (pruritis)
- In severe cases: uraemic pericarditis (friction rub on auscultation), seizures, coma
Symptoms pointing to the CAUSE:
- Dry mouth, postural dizziness, sunken eyes = dehydration (prerenal)
- Fever, rash, recent antibiotic = AIN (drug reaction)
- Inability to pass urine, enlarged prostate felt on PR exam = postrenal
- Recent crush injury or dark brown urine = rhabdomyolysis (myoglobinuria)
- Joint pain, rash, blood in cough = vasculitis/glomerulonephritis
5. Investigations - What to Order and Why
Blood Tests
| Test | Why you order it |
|---|---|
| Serum Creatinine + eGFR | Confirms AKI, stages it, monitors progress. Creatinine rises as filtration drops. |
| Blood Urea Nitrogen (BUN) | Also rises in AKI. BUN:Creatinine ratio >20:1 = classic prerenal pattern (tubules are concentrating and reabsorbing urea) |
| Electrolytes (K+, Na+, HCO3-) | AKI causes hyperkalaemia (K+ can't be excreted - dangerous, causes cardiac arrhythmias). Metabolic acidosis (bicarb falls). Hyponatraemia possible. |
| Full blood count | Anaemia (chronic), eosinophilia (suggests AIN or atheroemboli) |
| Serum calcium, phosphate | Hypercalcaemia can cause prerenal AKI. Hyperphosphataemia builds up as kidneys fail. |
| LDH, CK (Creatine Kinase) | Very high CK = rhabdomyolysis causing toxic ATN |
| LFTs + Albumin | Low albumin = hypoalbuminaemia suggesting nephrotic syndrome or liver disease. Liver disease can cause hepatorenal syndrome. |
| ABG (blood gas) | Checks for metabolic acidosis (low pH, low bicarb) |
| Blood cultures | If sepsis is suspected as the cause |
Urine Tests
| Test | Why you order it | What it tells you |
|---|---|---|
| Urine output measurement | Fundamental - oliguria is the hallmark | Confirms AKI severity |
| Urinalysis (dipstick) | Quick first-line screen | Blood = glomerulonephritis or stone; protein = glomerular disease; leucocytes + nitrites = infection causing AKI |
| Urine microscopy (urine sediment) | Most informative single urine test | Muddy brown granular casts = ATN (dead tubule cells); RBC casts = glomerulonephritis; WBC casts = AIN; Hyaline casts = bland, suggests prerenal; Waxy/broad casts = chronic disease |
| Spot urine Na+ and FENa (Fractional Excretion of Sodium) | Differentiates prerenal from ATN | FENa <1% = kidney trying to hold onto sodium = prerenal (or early obstruction); FENa >2% = tubules damaged, can't reabsorb = ATN |
| Urine osmolality | Supports prerenal vs ATN | Urine osm >500 mOsm/kg = concentrated = prerenal; <350 = dilute = ATN (damaged tubules can't concentrate) |
| Urine creatinine | Used to calculate FENa and spot creatinine:protein ratio | |
| Urine protein:creatinine ratio | Quantifies proteinuria | >3.5 g/g = nephrotic range (suggests glomerular disease) |
Imaging
| Test | Why you order it |
|---|---|
| Renal ultrasound | First-line imaging - essential in every AKI patient. Rules out obstruction (hydronephrosis = dilated collecting system = postrenal AKI). Shows kidney size (small kidneys = CKD, not AKI). Also shows stones, masses. |
| Bladder scan (post-void residual) | If BPH or neurogenic bladder suspected - checks for urinary retention |
| Chest X-ray | Detects pulmonary oedema (fluid in lungs) from fluid overload |
| ECG | If hyperkalaemia - peaked T waves, wide QRS, sine wave pattern = emergency |
| Renal Doppler | If renal artery stenosis or renal vein thrombosis suspected |
| CT scan | If stone or structural obstruction not seen on ultrasound |
| Renal biopsy | If cause unclear, or glomerulonephritis / vasculitis suspected (to guide immunosuppression treatment) |
6. Quick Summary of Confirming Clues
| Finding | What it confirms |
|---|---|
| Rising creatinine + oliguria | AKI is present (by definition) |
| BUN:Cr ratio >20:1, FENa <1%, concentrated urine | Prerenal AKI |
| Muddy brown granular casts in urine | ATN (intrinsic) |
| RBC casts + haematuria + proteinuria | Glomerulonephritis |
| Rash, eosinophilia, recent drug, WBC casts | AIN |
| Hydronephrosis on ultrasound + anuria | Postrenal obstruction |
| CK >10,000, dark urine, no RBCs on microscopy | Rhabdomyolysis causing ATN |
| Peaked T waves on ECG | Dangerous hyperkalaemia from AKI |
7. General Principles of Management
- Treat the cause - rehydrate for prerenal, relieve obstruction for postrenal, remove offending drug for AIN
- Monitor fluid balance strictly - daily weights, urine output
- Watch and treat hyperkalaemia - most dangerous electrolyte abnormality (can cause cardiac arrest)
- Avoid nephrotoxins - stop NSAIDs, aminoglycosides, contrast where possible
- Nutrition - adequate protein and calories
- Dialysis (renal replacement therapy) when:
- Refractory hyperkalaemia (K+ >6.5)
- Severe fluid overload not responding to diuretics
- Severe metabolic acidosis (pH <7.1)
- Uraemic encephalopathy or pericarditis
Sources: Frameworks for Internal Medicine, pp. 448-453; National Kidney Foundation Primer on Kidney Diseases 8e, pp. 347-350; Robbins & Kumar Basic Pathology, p. 2031; Brenner & Rector's The Kidney
What would be the patient complaint c/o ?
A patient with AKI typically comes in with these complaints, grouped by what's happening in the body:
Patient Complaints (Chief Complaints) in AKI
🟡 Urine-Related (Most Classic)
- "I'm passing very little urine" - oliguria, the most common presenting complaint
- "I haven't passed urine at all since yesterday" - anuria (severe or postrenal)
- "My urine looks dark / cola-colored / tea-colored" - suggests myoglobinuria (rhabdomyolysis) or haematuria
- "My urine looks foamy" - suggests proteinuria (glomerular disease)
🟠 Fluid Overload Complaints
- "My legs are swollen" - pedal oedema from fluid retention
- "My face is puffy especially in the morning" - periorbital oedema
- "I'm gaining weight rapidly" - water retention
- "I'm short of breath / breathless even at rest" - pulmonary oedema (fluid in lungs)
- "I can't lie flat, I need extra pillows" - orthopnoea from pulmonary oedema
🔴 Uraemic Symptoms (Waste Building Up)
- "I feel very weak and tired all the time" - most common non-specific complaint
- "I feel nauseated / I'm vomiting" - uraemic toxins irritating the gut
- "I have no appetite" - anorexia from uraemia
- "I have a headache / I feel confused" - uraemic encephalopathy
- "I have muscle cramps" - electrolyte imbalance (Ca²⁺, Mg²⁺)
- "I keep hiccupping" - classic uraemic hiccups
- "I'm itching all over" - pruritis from urea crystals deposited in skin
- "I feel chest pain" - uraemic pericarditis (scratchy, positional pain)
🟢 Complaints That Point to the Cause
| Complaint | Likely Cause |
|---|---|
| "I've had bad diarrhoea and vomiting for 3 days" | Dehydration → Prerenal |
| "I lost a lot of blood" | Hypovolaemia → Prerenal |
| "I've been taking painkillers (ibuprofen) a lot" | NSAIDs → Prerenal or AIN |
| "I can't start urinating, I feel my bladder is full" | Urinary retention → Postrenal (BPH) |
| "I had severe pain in my lower back/flank" | Kidney stone → Postrenal |
| "I had a crush injury / my muscles are very sore" | Rhabdomyolysis → ATN |
| "I started a new antibiotic recently, now I have a rash and fever" | Drug-induced AIN |
| "I've been very ill with fever / infection" | Sepsis → Prerenal or ATN |
🔵 Electrolyte-Related Complaints (Often Missed)
- "My heart is racing / fluttering" - hyperkalaemia causing arrhythmia
- "I feel weak in my muscles" - hyperkalaemia or acidosis
- "I feel dizzy when I stand up" - postural hypotension from dehydration (prerenal)
Simple Way to Remember
A patient with AKI walks in saying: "Doctor, I'm barely urinating, my legs are swollen, I'm feeling very weak and nauseous, and I've been vomiting for days"
- That's your classic presentation.
The single most common complaint is either decreased urine output or just weakness/fatigue - many patients don't even notice the urine change until you specifically ask.
Pro tip for history taking: Always ask these 3 targeted questions:
- "How much urine are you passing compared to normal?"
- "Any recent illness, blood loss, vomiting, diarrhoea, new medications?"
- "Any difficulty passing urine or feeling the bladder is full?"
These three questions alone can often point you straight to prerenal, intrinsic, or postrenal.
Explain me anaemia from basic to clinics but not in detail
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have all the information needed for a clear, well-structured simplified guide. Let me compose the answer.
Anaemia - From Basic to Clinical
1. What is Anaemia?
Anaemia is a significant reduction in the number of red blood cells (RBCs) or their haemoglobin content, resulting in reduced oxygen delivery to the body's tissues.
Think of it this way: blood is your oxygen delivery truck. Haemoglobin is the cargo (oxygen). Anaemia means either fewer trucks, or trucks carrying less oxygen.
WHO Definition (diagnosis threshold):
| Group | Hb level for anaemia |
|---|---|
| Adult men | < 13 g/dL |
| Adult women | < 12 g/dL |
| Pregnant women | < 11 g/dL |
| Children | varies by age |
2. Normal RBC Life Cycle (Quick Background)
- Bone marrow makes RBCs from stem cells (needs iron, B12, folate, EPO)
- RBC lives ~120 days in circulation carrying oxygen
- Old RBCs are broken down in the spleen and liver
- Iron is recycled back to bone marrow
Anaemia happens when this balance breaks - either less production, more destruction, or more loss.
3. Classification - 2 Ways to Think About It
Method 1 - By Red Cell Size (MCV on CBC) - Most Practical Clinically
The MCV (Mean Corpuscular Volume) tells you the size of the RBC.
| Type | MCV | Think of it as | Main Causes |
|---|---|---|---|
| Microcytic (small cells) | < 80 fL | Small trucks with less space | Iron deficiency, Thalassemia, Anaemia of chronic disease, Sideroblastic anaemia |
| Normocytic (normal size) | 80-100 fL | Normal trucks, just fewer | Blood loss (acute), Anaemia of chronic disease, Renal failure, Aplastic anaemia, Haemolysis (early) |
| Macrocytic (large cells) | > 100 fL | Oversized trucks that don't work well | B12/Folate deficiency, Alcohol, Liver disease, Hypothyroidism, Drugs (methotrexate) |
Method 2 - By Mechanism (More Conceptual)
| Mechanism | What's happening | Examples |
|---|---|---|
| Underproduction | Bone marrow not making enough RBCs | Iron deficiency, B12/folate deficiency, aplastic anaemia, renal anaemia |
| Increased destruction (Haemolysis) | RBCs being destroyed too fast | Sickle cell, G6PD deficiency, autoimmune haemolytic anaemia, malaria, TTP/HUS |
| Blood loss | Losing RBCs externally | GI bleed, trauma, heavy periods, surgery |
4. Key Types - Simplified
🔴 Iron Deficiency Anaemia (most common worldwide)
- Cause: Poor intake, chronic blood loss (GI bleed, heavy menstruation), malabsorption (coeliac disease)
- RBC: Microcytic, hypochromic (small, pale cells)
- Specific signs: Koilonychia (spoon-shaped nails), angular stomatitis (cracks at mouth corners), glossitis (sore tongue), pica (craving non-food items like clay/ice)
- Ferritin: Low (most sensitive marker)
🔵 B12 / Folate Deficiency Anaemia (Megaloblastic)
- Cause: Poor diet, veganism (B12), alcohol (folate), malabsorption (pernicious anaemia - no intrinsic factor for B12)
- RBC: Macrocytic, oval-shaped; hypersegmented neutrophils on smear (classic clue)
- Specific signs of B12 deficiency: Subacute combined degeneration of spinal cord - numbness, tingling, balance problems, psychiatric symptoms. Folate deficiency does NOT cause this.
🟢 Anaemia of Chronic Disease (ACD)
- Cause: Chronic inflammation, infection, cancer, CKD - inflammatory cytokines block iron release and reduce EPO response
- RBC: Normocytic (or mildly microcytic)
- Key point: Ferritin is normal or high (iron is trapped inside cells, not actually deficient). This distinguishes it from iron deficiency.
🟡 Haemolytic Anaemia
- Cause: RBCs being destroyed early. Can be hereditary (sickle cell, G6PD, spherocytosis) or acquired (autoimmune, malaria, TTP)
- RBC: Normocytic usually; reticulocytes high (bone marrow working overtime)
- Specific signs: Jaundice (bilirubin from destroyed Hb), dark urine (urobilinogen), splenomegaly
- Labs: Raised LDH, raised unconjugated bilirubin, low haptoglobin
⚫ Aplastic Anaemia
- Cause: Bone marrow failure - stops making all blood cells (RBCs, WBCs, platelets)
- RBC: Normocytic; pancytopenia (all three cell lines low)
- Presentation: Anaemia + infections (low WBCs) + bleeding (low platelets)
5. Symptoms - What Patients Feel
All types of anaemia share common symptoms from reduced oxygen delivery:
General (from hypoxia):
- Fatigue and weakness (most common, earliest symptom)
- Shortness of breath on exertion
- Dizziness / light-headedness
- Headache
- Poor concentration
Cardiovascular (compensation):
- Palpitations (heart beating faster to compensate)
- Tachycardia on examination
- In severe or elderly patients: chest pain (angina), heart failure symptoms
On examination:
- Pallor - look at: conjunctiva (inner lower eyelid), palmar creases, nail beds, tongue
- Tachycardia at rest
- Ejection systolic murmur - high flow murmur from hyperdynamic circulation (not a structural heart problem)
Important: Symptoms depend on how fast anaemia develops. Slow anaemia (chronic) is much better tolerated than sudden anaemia (acute blood loss) because the body adapts with increased cardiac output, increased 2,3-DPG (shifts oxygen curve), and plasma expansion.
6. Investigations - What to Order and Why
First-line (always)
| Test | Why | What it tells you |
|---|---|---|
| Complete Blood Count (CBC/FBC) | The essential first test | Hb level, MCV (size), RBC count, WBC, platelets |
| Peripheral blood smear | Cheap, hugely informative | Confirms cell morphology - microcytes, macrocytes, sickle cells, schistocytes, hypersegmented neutrophils |
| Reticulocyte count | Tells you if marrow is responding | High reticulocytes = blood loss or haemolysis (marrow working); Low = underproduction (marrow not working) |
Second-line (based on MCV type)
If MICROCYTIC:
| Test | Why |
|---|---|
| Serum Ferritin | Best marker for iron stores. Low = iron deficiency |
| Serum Iron + TIBC | Iron low + TIBC high = iron deficiency. Iron low + TIBC low/normal = ACD |
| Haemoglobin electrophoresis | If thalassaemia suspected |
If MACROCYTIC:
| Test | Why |
|---|---|
| Serum B12 level | Low in B12 deficiency (pernicious anaemia, vegans) |
| Serum/RBC Folate | Low in folate deficiency (alcohol, poor diet, pregnancy) |
| LFTs + TFTs | Liver disease and hypothyroidism cause macrocytic anaemia |
| Anti-intrinsic factor antibodies | Confirms pernicious anaemia |
If NORMOCYTIC or suspected haemolysis:
| Test | Why |
|---|---|
| LDH + Bilirubin (indirect) | Both raised in haemolysis |
| Haptoglobin | Low/absent in haemolysis (haptoglobin mops up free Hb) |
| Direct Coombs Test (DAT) | Positive in autoimmune haemolytic anaemia |
| G6PD assay | If G6PD deficiency suspected |
| Renal function (urea/creatinine) | CKD causes normocytic anaemia (low EPO) |
| Bone marrow biopsy | If aplastic anaemia or marrow infiltration suspected |
7. Quick "How to Approach Anaemia" Flowchart
Low Hb confirmed on CBC
↓
Look at MCV
↙ ↓ ↘
Micro Normo Macro
↓ ↓ ↓
Ferritin Retic B12/Folate
Low→IDA High→ Low→Megaloblastic
Haem/ Normal→Alcohol/
Hb elec Bleed Liver/Hypothyroid
→Thal Low→
Underproduction
(CKD, aplastic)
8. Simple Memory Aid
"FITS" for microcytic: Ferritin↓ (IDA), Iron block (ACD), Thalassaemia, Sideroblastic"BFALMs" for macrocytic: B12, Folate, Alcohol, Liver disease, Medications, hypothyroidiSmNormocytic = think chronic disease, CKD, haemolysis, bone marrow problem
Sources: Harrison's Principles of Internal Medicine 22e, pp. 491-492; Goldman-Cecil Medicine, p. 1666; Symptom to Diagnosis, 4th ed., pp. 9560-9615
Teach me sickle cell anaemia from basic to clinics but not in too deep
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Excellent - I have all the content and images I need. Here is the full, simplified guide.
Sickle Cell Anaemia - From Basic to Clinical
1. What Is It?
Sickle cell anaemia is a hereditary (genetic) disease where a single DNA mutation causes the haemoglobin inside red blood cells to be abnormal. This abnormal haemoglobin is called HbS (haemoglobin S).
The result: red blood cells that are normally round and flexible become rigid, sticky, and sickle (crescent) shaped when they lose oxygen. These abnormal cells cause two big problems:
- They break down too fast (haemolytic anaemia)
- They block small blood vessels (vaso-occlusion → organ damage + pain)
2. The Genetics - Simple Version
Normal haemoglobin = HbA (two alpha chains + two beta chains)
The mutation is in the beta-globin gene on chromosome 11:
- One base change (A→T) causes glutamate (acidic, water-loving) to be replaced by valine (hydrophobic) at position 6 of the beta chain
- This tiny change makes HbS stick together when deoxygenated
Inheritance Pattern (Autosomal Recessive):
| Genotype | What they have | Condition |
|---|---|---|
| HbA/HbA | Two normal genes | Normal |
| HbA/HbS | One normal + one sickle gene | Sickle Cell TRAIT - carrier, mostly healthy |
| HbS/HbS | Two sickle genes | Sickle Cell DISEASE - full disease |
Sickle cell trait (HbA/HbS): only ~40% of Hb is HbS. HbA dilutes it out and prevents sickling under normal conditions. These people are usually healthy but can sickle in extreme hypoxia (e.g., unpressurized aircraft, high altitude).
Why is it so common?
HbS protects against falciparum malaria - so the gene survived by natural selection in malaria-endemic areas: sub-Saharan Africa, Mediterranean, Middle East, India. ~8% of African-Americans are carriers.
3. How Does the Cell Actually Sickle? (Pathophysiology)
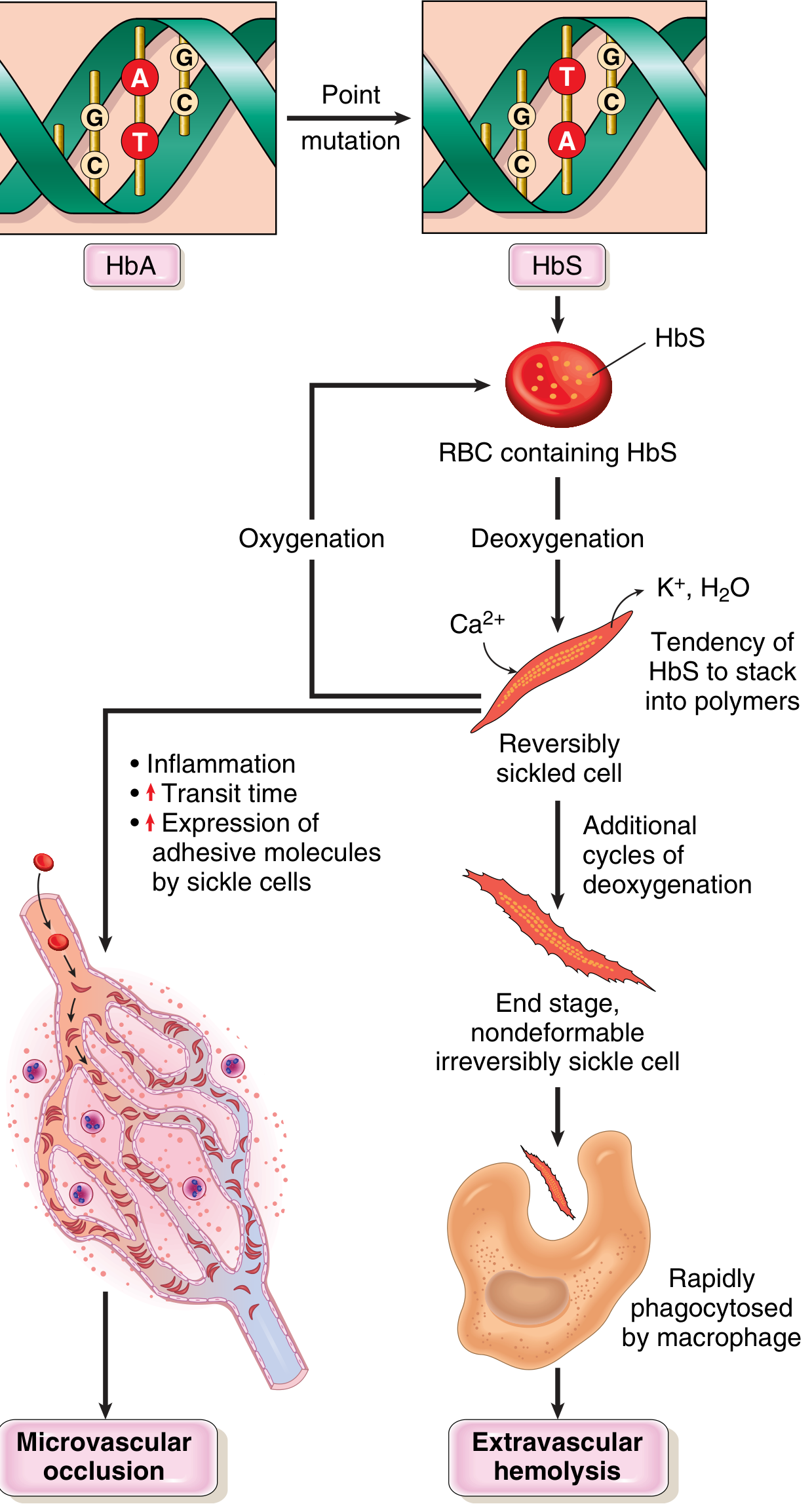
Step by step:
- When the RBC loses oxygen (in peripheral tissues), HbS molecules change shape
- They stack into long rigid polymers (chains) inside the cell
- This distorts the cell into a sickle/crescent shape
- If the cell gets reoxygenated - it can go back to normal (reversible sickling)
- But repeated cycles of sickling cause permanent membrane damage (Ca²⁺ enters, K⁺ and water leave)
- Eventually: irreversibly sickled cells that cannot return to normal
- These cells are either:
- Destroyed by macrophages in the spleen → haemolytic anaemia
- Stuck in small blood vessels → block blood flow → tissue ischaemia → pain crisis + organ damage
Triggers that make sickling worse:
- Low oxygen (hypoxia)
- Dehydration (concentrates HbS)
- Infection / inflammation (slows blood flow, activates endothelium)
- Cold temperatures
- Acidosis
- Strenuous exercise
4. What Do Sickle Cells Look Like? (Blood Smear)
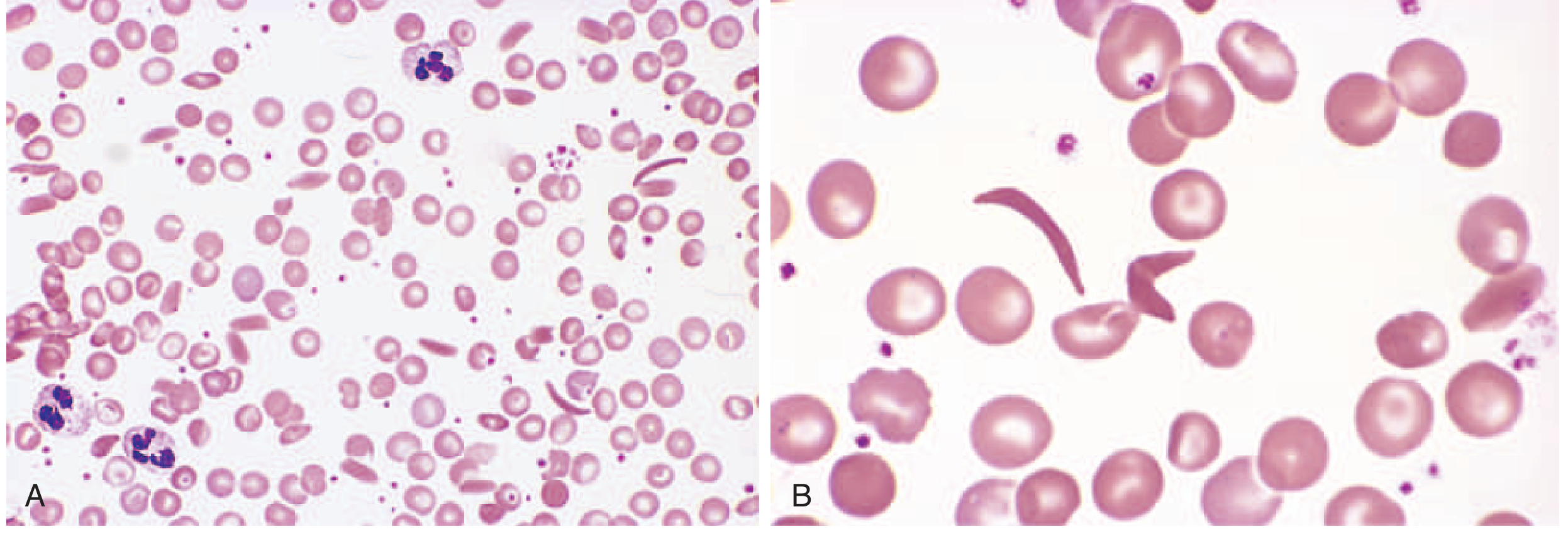
On blood smear: elongated, crescent/boat-shaped cells alongside normal round RBCs, target cells, and anisocytosis (variable cell sizes).
5. Why Doesn't the Baby Get Sick at Birth?
Newborns have Foetal Haemoglobin (HbF, α₂γ₂) which does NOT sickle and actually INHIBITS HbS polymerisation. Symptoms only appear when HbF is replaced by HbS - usually around 5-6 months of age.
This is also why HbF is therapeutically important (hydroxyurea raises HbF levels - see treatment below).
6. Clinical Features - What Happens to the Patient?
Background (always present):
- Chronic haemolytic anaemia - Hb usually 6-9 g/dL (normal 12-16)
- Jaundice - from constant RBC breakdown releasing bilirubin
- Pigment gallstones - excess bilirubin in bile
- Reticulocytosis - bone marrow working overtime to replace destroyed cells
CRISES - the dramatic episodes:
1. Vaso-occlusive (Pain) Crisis - Most Common
- Sickle cells block small blood vessels → ischaemia and infarction in bones, muscles, organs
- Severe pain - bones (back, chest, limbs), abdomen
- Triggered by infection, dehydration, cold, stress
- Hand-foot syndrome (dactylitis) - swollen, painful hands and feet from bone infarction - often the first sign in infants
2. Acute Chest Syndrome - Most Dangerous
- Sickling inside the lung blood vessels
- Presents like pneumonia: fever, chest pain, falling oxygen levels, new lung infiltrate on X-ray
- Creates a vicious cycle: lung sickling → more hypoxia → more sickling
- Leading cause of death in SCD patients
3. Stroke
- Sickling in cerebral blood vessels
- Common in children; can cause sudden hemiplegia, seizures, coma
- Second leading cause of death after acute chest syndrome
4. Aplastic Crisis
- Sudden stop in RBC production
- Triggered by parvovirus B19 infection (infects erythroid precursors in bone marrow)
- Hb drops suddenly and severely - very dangerous but self-limited (2-3 weeks)
5. Sequestration Crisis (mainly in young children)
- Large amounts of blood suddenly trapped in the spleen
- Rapid splenomegaly, fall in Hb, cardiovascular collapse
- Medical emergency
Organ Damage Over Time (chronic complications):
| Organ | What Happens | Result |
|---|---|---|
| Spleen | Repeated infarctions → auto-splenectomy | No spleen function by adulthood → susceptible to encapsulated bacteria |
| Kidneys | Papillary necrosis, proteinuria | Renal failure |
| Eyes | Proliferative retinopathy | Visual loss, blindness |
| Bones | Avascular necrosis | Hip/shoulder destruction |
| Brain | Silent infarcts | Cognitive problems, strokes |
| Skin | Poor blood flow | Chronic leg ulcers |
| Penis | Vascular stasis | Priapism → erectile dysfunction |
| Heart | Compensatory cardiomegaly | Cardiomyopathy |
Infections - Major Problem:
Functional asplenia makes patients vulnerable to encapsulated bacteria:
- Streptococcus pneumoniae
- Haemophilus influenzae
- Neisseria meningitidis
Also prone to Salmonella osteomyelitis (most common cause of bone infection in SCD - not Staphylococcus as in normal patients).
7. Diagnosis
| Test | Finding | Why |
|---|---|---|
| Full Blood Count (FBC) | Hb 6-9 g/dL, normocytic, high reticulocyte count | Chronic haemolytic anaemia |
| Peripheral blood smear | Sickle cells, target cells, Howell-Jolly bodies (asplenia) | Morphological diagnosis |
| Haemoglobin electrophoresis | Gold standard - shows HbS band, absent HbA | Confirms diagnosis and genotype |
| Newborn screening (heel-prick) | Identifies HbS at birth | Mandated in many countries |
| Sickle solubility test | HbS precipitates in reducing solution | Screening tool (cannot differentiate trait from disease) |
| Bilirubin + LDH | Raised | Haemolysis markers |
| Reticulocyte count | High (>5-10%) | Compensatory RBC production |
| Prenatal diagnosis | Amniocentesis or chorionic villus sampling → DNA analysis | For at-risk families |
8. Treatment - Simplified
Long-term / Preventive:
| Treatment | Why |
|---|---|
| Hydroxyurea (main drug) | Raises HbF levels → inhibits sickling. Also reduces WBC (anti-inflammatory) and increases RBC size (dilutes HbS). Reduces pain crises by ~50% |
| Penicillin prophylaxis (in children <5 yrs) | Prevents fatal pneumococcal sepsis from functional asplenia |
| Vaccinations | Pneumococcal, meningococcal, Hib - essential due to asplenia |
| Folic acid | Supports bone marrow in making new RBCs |
| Regular blood transfusions | For stroke prevention (in high-risk children), severe anaemia |
During Crisis:
| Treatment | Why |
|---|---|
| IV fluids / oral hydration | Treats dehydration (a key trigger), improves blood flow |
| Analgesia (paracetamol → NSAIDs → opioids) | Pain management is priority |
| Oxygen | Reduces hypoxia-driven sickling |
| Treat the trigger | Antibiotics for infection, etc. |
| Exchange transfusion | For acute chest syndrome, stroke - rapidly lowers % HbS |
Curative:
- Allogeneic bone marrow (stem cell) transplant - only current cure; best in children with severe disease and a matched sibling donor
- Gene therapy - promising; trials showing correction of the HbS mutation or induction of HbF permanently
9. Prognosis
- 50% of patients now survive beyond the 5th decade with good care
- Most common causes of death: acute chest syndrome and stroke
- Clinical course is highly variable - some patients have frequent crises, others go years between events
Quick Summary - The Core Concept
One amino acid change (Glu → Val) in the beta-globin gene makes haemoglobin sticky when deoxygenated. This turns round flexible RBCs into rigid sickle-shaped cells that block vessels and get destroyed early. The result: chronic anaemia + episodic pain crises + progressive organ damage due to ischaemia.
Sources: Robbins & Kumar Basic Pathology, pp. 386-388; Harrison's Principles of Internal Medicine 22e; Rosen's Emergency Medicine 10e
Thalassemia from basic to clinics with proper protocol and management with proper diagnosis
Finding Sources
Finding Sources
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.