Explain hyperkalemia
Reading File
Finding Sources
Searching PubMed
hyperkalemia management treatment
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Hyperkalemia
Hyperkalemia is defined as a serum potassium (K⁺) concentration above 5.5 mEq/L (some sources use >5.0 mEq/L). It represents an excess of potassium in the extracellular fluid and is potentially one of the most rapidly lethal electrolyte disorders, primarily due to its effects on cardiac conduction.
Potassium Homeostasis - Background
Normal serum K⁺ is maintained in the narrow range of 3.5-5.0 mEq/L by the kidneys, regulated via the renin-angiotensin-aldosterone (RAA) system. Approximately 98% of total body potassium is intracellular, making transcellular shifts the primary driver of acute changes in serum levels. Aldosterone promotes potassium secretion in the distal tubule; insulin and β2-adrenergic stimulation shift K⁺ intracellularly. Metabolic acidosis causes K⁺ efflux from cells in exchange for H⁺ ions. - Medical Physiology, p. 1196 / Current Surgical Therapy 14e, p. 3133
Epidemiology
Hyperkalemia occurs in 1.1%-10% of all hospitalized patients, with ~1% having significant hyperkalemia (K⁺ ≥6.0 mmol/L). It carries a mortality rate of 14.3%-41% in affected inpatients. Its prevalence rises sharply as GFR falls - from 2% at GFR 60-90 mL/min to 42% at GFR <20 mL/min. Renal failure is the most common cause presenting to the ED. - Brenner & Rector's The Kidney, p. 766
Causes
1. Pseudohyperkalemia (Factitious)
Before assuming true hyperkalemia, rule out lab artifact:
- Traumatic hemolysis during blood drawing (most common cause of a "high" reported lab value)
- Fist clenching / tourniquet use causing local K⁺ efflux
- Thrombocytosis, leukocytosis, or erythrocytosis - cellular K⁺ released during clot formation in the sample tube
- Acute anxiety-induced respiratory alkalosis with redistribution
2. Increased Potassium Load
- Large exogenous IV K⁺ bolus (iatrogenic - wrong IV fluid)
- High dietary intake (especially with impaired renal excretion)
- Massive cell breakdown: intravascular hemolysis (mismatched transfusion), crush injuries, burns, rhabdomyolysis, tumor lysis syndrome, GI bleeding with intestinal reabsorption
- Chemotherapy-induced cell death
3. Transcellular Redistribution (K⁺ shifts out of cells)
- Metabolic acidosis (non-organic - inorganic acidosis causes larger shift than organic)
- Insulin deficiency (insulin is required for cellular K⁺ uptake)
- β-adrenergic blockade (inhibits Na-K-ATPase and cellular uptake)
- Digitalis toxicity (inhibits Na-K-ATPase pump)
- Succinylcholine - depolarizes muscle, releasing K⁺; the risk is greatly amplified in denervated muscle (burns, crush injuries, prolonged immobility)
- Hypertonicity (e.g., mannitol, hyperglycemia) - water draws out K⁺ by solvent drag
- Hyperkalemic periodic paralysis
- Drugs opening KATP channels (cyclosporine, nicorandil)
4. Impaired Renal Excretion (most common cause of sustained hyperkalemia)
-
Advanced renal failure (any cause - GFR <20 mL/min)
-
Hypoaldosteronism - primary adrenal insufficiency, isolated aldosterone deficiency, hyporeninemic hypoaldosteronism (diabetic nephropathy, NSAIDs, ACE inhibitors, heparin)
-
Medications blocking the RAA axis: ACE inhibitors, ARBs, spironolactone, eplerenone, amiloride, triamterene, NSAIDs, trimethoprim, pentamidine, calcineurin inhibitors
-
Decreased distal Na⁺ delivery (limits K⁺ secretion)
-
Tubulointerstitial diseases, urinary obstruction, sickle cell disease, SLE
-
Medical Physiology, p. 1196 | Brenner & Rector's The Kidney | Current Surgical Therapy 14e
Clinical Manifestations
Hyperkalemia is clinically silent until major cardiotoxicity develops - this is what makes it dangerous.
Cardiac (most important)
The cardiac effects result from delayed depolarization and reduced membrane excitability. ECG changes progress in sequence with rising K⁺:
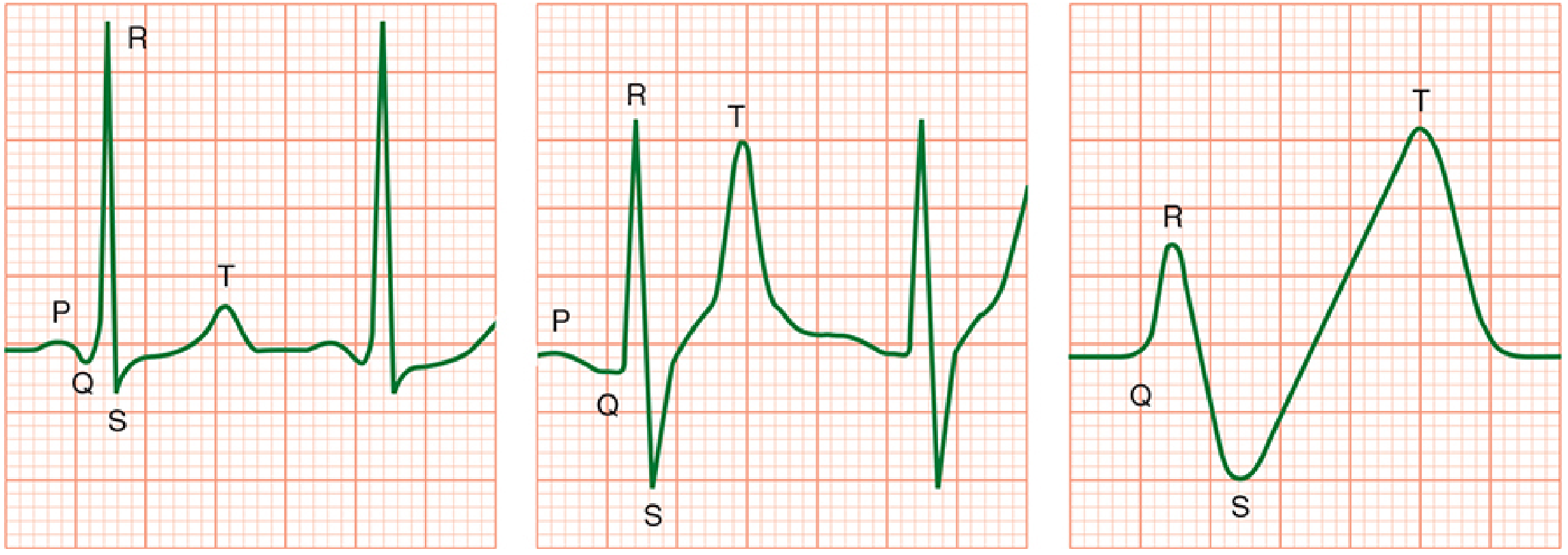
Progression of ECG changes in hyperkalemia - from normal, to peaked T waves with widened QRS, to the "sine wave" pre-arrest pattern. (Morgan & Mikhail's Clinical Anesthesiology)
| Sequence | ECG Finding | Approximate K⁺ level |
|---|---|---|
| 1st | Symmetrically peaked (tall, narrow) T waves + shortened QT | ~5.5-6.5 mEq/L |
| 2nd | Widening of QRS complex | ~6.5-7.0 mEq/L |
| 3rd | PR interval prolongation | ~7.0 mEq/L |
| 4th | Loss of P wave, loss of R-wave amplitude | >7.0 mEq/L |
| 5th | ST depression (or elevation) | >7.5 mEq/L |
| 6th | Sine wave pattern | >8.0 mEq/L |
| Terminal | Ventricular fibrillation / asystole | >8.0-9.0 mEq/L |
Note: ECG changes correlate only roughly with K⁺ level. A normal ECG does not exclude dangerous hyperkalemia - get a serum level. Hypocalcemia, hyponatremia, and acidosis all worsen cardiac effects.
Neuromuscular
- Skeletal muscle weakness generally not seen until K⁺ >8 mEq/L
- Can progress to flaccid paralysis (ascending)
Contractility
Preserved until late in the course - cardiac output may be relatively maintained even with severe ECG changes.
Diagnosis & Workup
- Rule out pseudohyperkalemia - repeat K⁺ in non-hemolyzed sample
- 12-lead ECG immediately - do not wait for labs if suspected
- Serum electrolytes, creatinine, BUN, osmolality, CBC, glucose, Mg²⁺, Ca²⁺
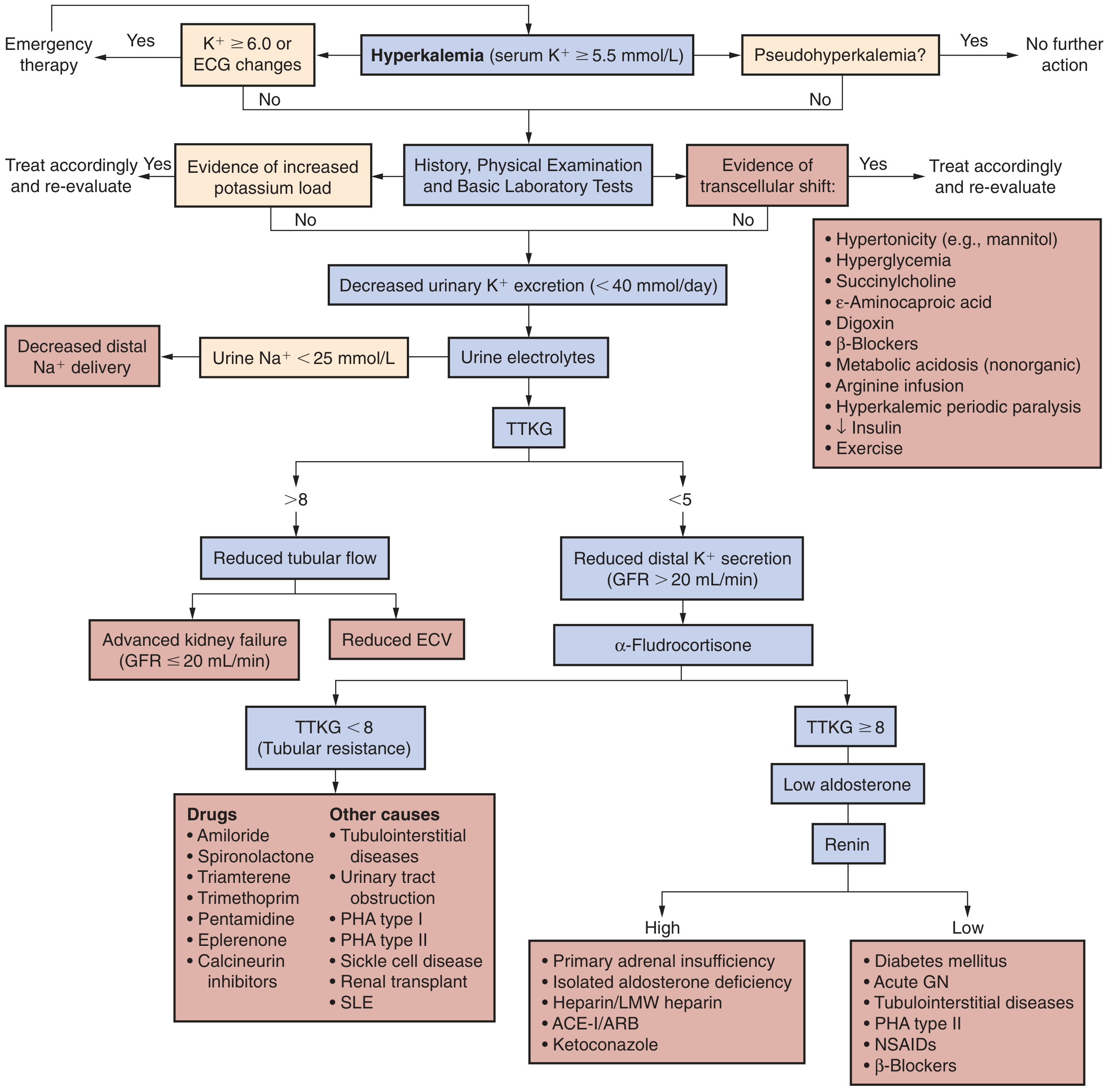
- Urine electrolytes (Na⁺, K⁺, osmolality, creatinine) for TTKG (transtubular K⁺ gradient) or urine K⁺/creatinine ratio
- Plasma renin activity and aldosterone if cause unclear
- History: medications (ACE-I, ARBs, NSAIDs, K⁺-sparing diuretics), diet, salt substitutes, urine output changes
The clinical approach flowchart below guides workup based on urinary K⁺ excretion, TTKG, aldosterone, and renin:
Treatment
Treatment follows three sequential goals: (1) stabilize the cardiac membrane, (2) shift K⁺ into cells, (3) remove K⁺ from the body.
Step 1 - Cardiac Membrane Stabilization (Fastest, does NOT lower K⁺)
| Agent | Dose | Onset | Duration | Mechanism |
|---|---|---|---|---|
| Calcium gluconate 10% | 10 mL IV over 2-3 min (repeat in 5 min if no response) | 1-3 min | 30-60 min | Raises action potential threshold; restores 15-mV resting-to-threshold potential gap |
| Calcium chloride 10% | 3-5 mL IV (via central line only - causes tissue necrosis if extravasated) | 1-3 min | 30-60 min | Same mechanism; 3x more elemental Ca²⁺ per mL |
Caution with digoxin: Hypercalcemia potentiates digoxin toxicity. If the patient is on digoxin, dilute calcium gluconate in 100 mL D5W and infuse over 20-30 min.
Step 2 - Redistribution into Cells (Temporary, buys time)
| Agent | Dose | Onset | Duration | Mechanism |
|---|---|---|---|---|
| Insulin + Glucose | 10 U regular insulin IV + 50 mL 50% dextrose (or 250 mL 10% dextrose) | ~30 min | 4-6 hrs | Stimulates Na-K-ATPase; shifts K⁺ intracellularly |
| Inhaled albuterol (β₂-agonist) | 10-20 mg nebulized (adult); 2.5-10 mg in children | 15-30 min | 2+ hrs | β₂ activation → Na-K-ATPase stimulation |
| Sodium bicarbonate | 50-100 mEq IV | 15-30 min | Variable | Alkalinization promotes cellular K⁺ uptake - less effective than previously thought; use with caution (volume overload, hypocalcemic tetany risk in renal failure) |
β-agonists and insulin/glucose effects are additive. Bicarbonate is a distant third choice.
Step 3 - Removal of K⁺ from the Body (Definitive)
| Agent | Route | Notes |
|---|---|---|
| Loop diuretics (furosemide) | IV | Effective if residual renal function present; use slow infusion (not bolus) to avoid ototoxicity |
| Patiromer | Oral | Novel K⁺ binder; effective for chronic management, NOT for acute hyperkalemia |
| Sodium zirconium cyclosilicate (ZS-9) | Oral | Newer non-absorbable cation exchanger; used in CKD/HF |
| Sodium polystyrene sulfonate (Kayexalate) | Oral/rectal | Older resin; no longer considered reliably effective; sorbitol carrier carries risk of bowel ischemia; contraindicated in surgical obstruction |
| Hemodialysis | - | Definitive treatment for severe hyperkalemia, especially in ESKD or when other measures fail |
Severity-Based Decision Framework
| Severity | K⁺ Level | ECG | Management |
|---|---|---|---|
| Mild | 5.5-6.0 mEq/L | None | Observe; eliminate K⁺ sources; dietary restriction (≤60 mEq/day); adjust medications |
| Moderate | 6.0-6.5 mEq/L | Peaked T waves only | Shift (insulin/glucose ± albuterol); add binder or diuretic; monitor ECG continuously |
| Severe | ≥6.5 mEq/L or any ECG changes beyond peaked T waves | QRS widening or worse | Medical emergency: Calcium IV first → redistribution → K⁺ removal; admit + continuous ECG monitoring |
| Critical | ≥8.0 mEq/L or sine wave/arrest | Sine wave, VF | Immediate calcium + redistribution + emergent dialysis; if cardiac arrest, standard ACLS + empirical treatment |
Special Situations
- CKD patients: Even modest K⁺ loads (dietary, from drugs like β-blockers or ACE-I) can precipitate severe hyperkalemia. A K⁺ of 6 mEq/L is potentially dangerous even without ECG changes. Succinylcholine should be used with caution (though true ESKD without denervation is lower risk than previously thought).
- Cardiac arrest with CKD: Assume hyperkalemia and treat empirically during resuscitation.
- Succinylcholine + denervated muscle (burns, crush, immobility): AVOID - causes massive, exaggerated K⁺ efflux.
- Combination RAAS blockade: ACE-I + ARB + mineralocorticoid antagonist together greatly increases hyperkalemia risk, especially in CKD stage IV-V (eGFR <30). Maximum spironolactone dose with ACE-I/ARB is 25 mg/day.
- Novel K⁺ binders (patiromer, ZS-9): A 2025 systematic review and network meta-analysis confirms their efficacy for chronic hyperkalemia management in CKD and heart failure, particularly enabling continued RAAS inhibitor therapy.
Key Takeaways
- Hyperkalemia is clinically silent until life-threatening cardiac events occur - always check ECG and serum K⁺ proactively in at-risk patients (CKD, RAAS drugs, diabetes).
- The ECG is useful but imperfect - normal ECG does not exclude dangerous hyperkalemia.
- Calcium is always the first emergency drug - it stabilizes membranes without affecting K⁺ level.
- Insulin/glucose and albuterol are the fastest ways to lower serum K⁺, but only temporarily.
- Dialysis is the only reliable definitive treatment in ESKD or severe cases.
- K⁺ ≥6.5 mEq/L or any ECG change beyond peaked T waves = medical emergency.
- Medical Physiology (Boron & Boulpaep) | Brenner & Rector's The Kidney | Morgan & Mikhail's Clinical Anesthesiology | Rosen's Emergency Medicine | Current Surgical Therapy 14e
Wilson’s disease
Reading File
Finding Sources
Searching PubMed
"Wilson disease" diagnosis treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, multi-source content with excellent images. Let me compile the full answer.
Wilson's Disease (Hepatolenticular Degeneration)
Wilson's disease is an autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene, resulting in toxic copper accumulation primarily in the liver, brain, and eyes. If untreated, it is progressive and fatal.
Genetics & Epidemiology
- Gene: ATP7B on chromosome 13 - encodes a copper-transporting P-type ATPase expressed in hepatocytes
- Inheritance: Autosomal recessive; >500 known disease-specific mutations; most patients are compound heterozygotes (different mutations on each allele)
- Incidence: ~1 in 30,000-40,000 live births; carrier prevalence 1 in 90-180
- Age of onset: Usually between 6 and 40 years; can rarely present in the 5th-6th decade. Liver disease predominates in younger patients (<30 years); neuropsychiatric disease typically appears after age 20.
Pathobiology
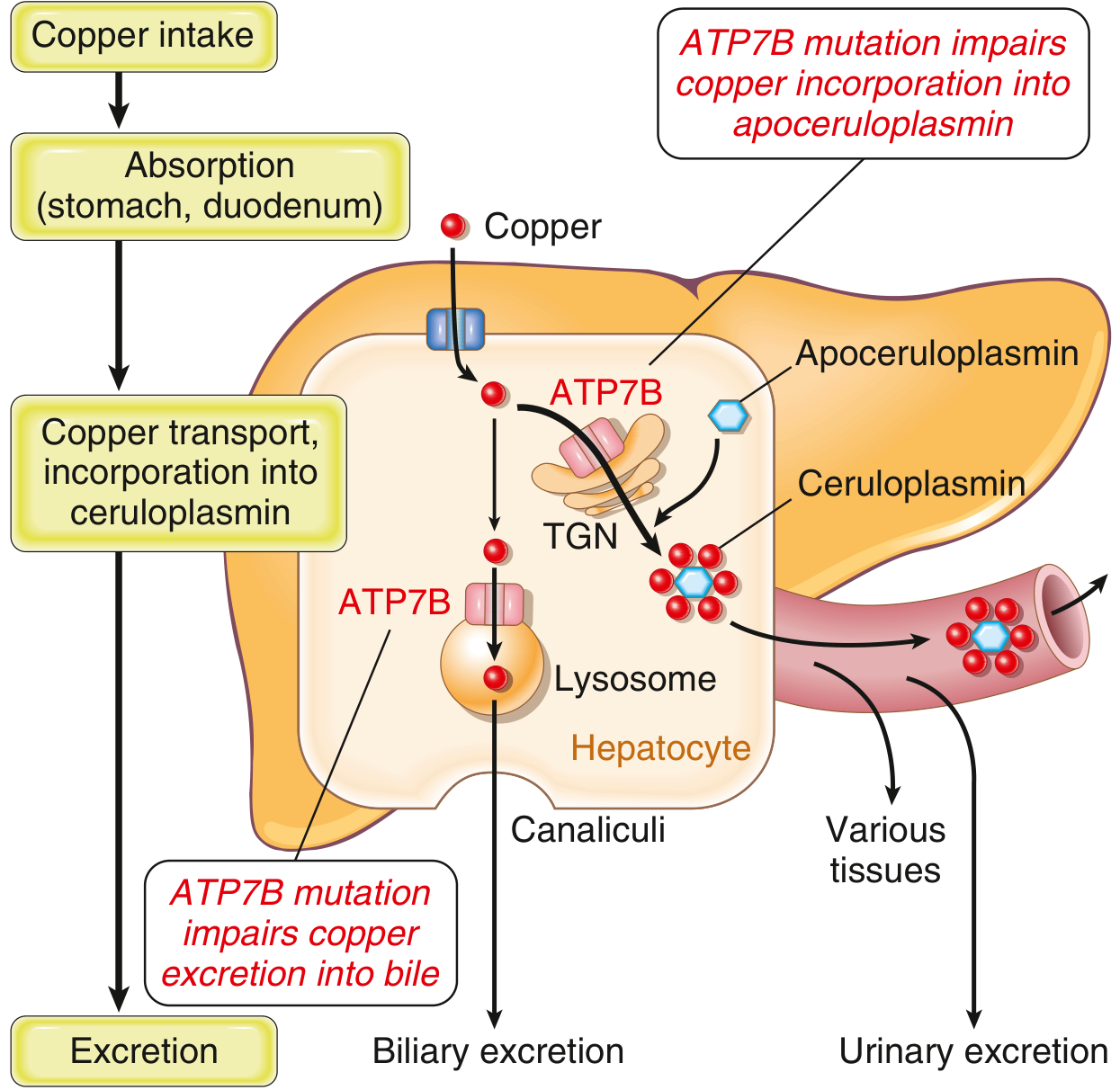
Normally, 40-60% of ingested copper (2-5 mg/day) is absorbed in the duodenum and proximal small intestine and transported to the liver bound to albumin. In the liver, ATP7B performs two functions:
- Trans-Golgi network: Transfers copper into apoceruloplasmin → forms ceruloplasmin, secreted into blood
- Lysosomes: Transports non-ceruloplasmin copper into bile canaliculi for fecal excretion (the primary excretion route; biliary copper does not undergo enterohepatic recirculation)
In Wilson disease, both pathways are impaired:
- Copper accumulates in hepatocyte cytoplasm and lysosomes → generates reactive oxygen species (ROS) → hepatocyte injury
- Ceruloplasmin synthesis is reduced (apoceruloplasmin is unstable without copper) → low serum ceruloplasmin (note: the low ceruloplasmin itself does not cause disease - it is a consequence, not a cause)
- As copper overloads the liver, non-ceruloplasmin-bound ("free") copper spills into circulation → hemolysis, deposits in brain, cornea, kidneys, bones, joints, parathyroid glands
- Urine copper excretion increases markedly
Because ceruloplasmin carries 90% of circulating copper, total serum copper is paradoxically low in most patients despite massive hepatic copper overload. - Goldman-Cecil Medicine, Robbins & Kumar Basic Pathology
Clinical Manifestations
About one-third of patients present with each: hepatic, neurologic, and psychiatric features. Some degree of liver disease is invariably present regardless of presenting complaint.
1. Hepatic (more common in younger patients)
| Presentation | Features |
|---|---|
| Asymptomatic / incidental | Elevated transaminases on routine testing |
| Steatohepatitis | Fatty change + Mallory hyaline, similar to NASH |
| Acute hepatitis | Mimics viral or autoimmune hepatitis |
| Acute liver failure (ALF) | ~5% of cases; presents with jaundice, coagulopathy, encephalopathy, Coombs-negative hemolytic anemia (from copper release), very low alkaline phosphatase |
| Chronic hepatitis / cirrhosis | Fatigue, jaundice, ascites, hepatomegaly, portal hypertension |
ALF Clue: In Wilson disease ALF, the alkaline phosphatase/total bilirubin ratio is <4 and AST/ALT ratio is >2.2 - these ratios accurately distinguish it from other causes of ALF. - Sleisenger & Fordtran's GI and Liver Disease
2. Neurological (typically presents after age 20)
Wilson disease is primarily a movement disorder (basal ganglia copper deposition). Features include:
- Dysarthria - often the first sign
- Dystonia (including sardonic "fixed smile")
- Tremor - can be postural, action, or "wing-beating" (flapping at the shoulders)
- Rigidity
- Chorea / choreiform movements
- Ataxia, gait disturbance
- Dysphagia
- Seizures (minority)
MRI shows abnormal T2 signal in the putamen (most characteristic), midbrain, pons, thalamus, and cerebellum. Cortical atrophy is common. - Bradley & Daroff's Neurology in Clinical Practice
3. Psychiatric (very common - ~50% of patients)
- Personality changes: irritability, anger, poor impulse control (most common)
- Depression (~30%)
- Anxiety
- Bipolar spectrum symptoms (~20%)
- Psychosis
- Suicidal ideation (5-15%)
- Frontosubcortical cognitive dysfunction
Wilson disease should be formally excluded in all young adults with new-onset psychiatric symptoms, especially if liver function tests are abnormal or there is a family history.
4. Ocular - Kayser-Fleischer (KF) Rings
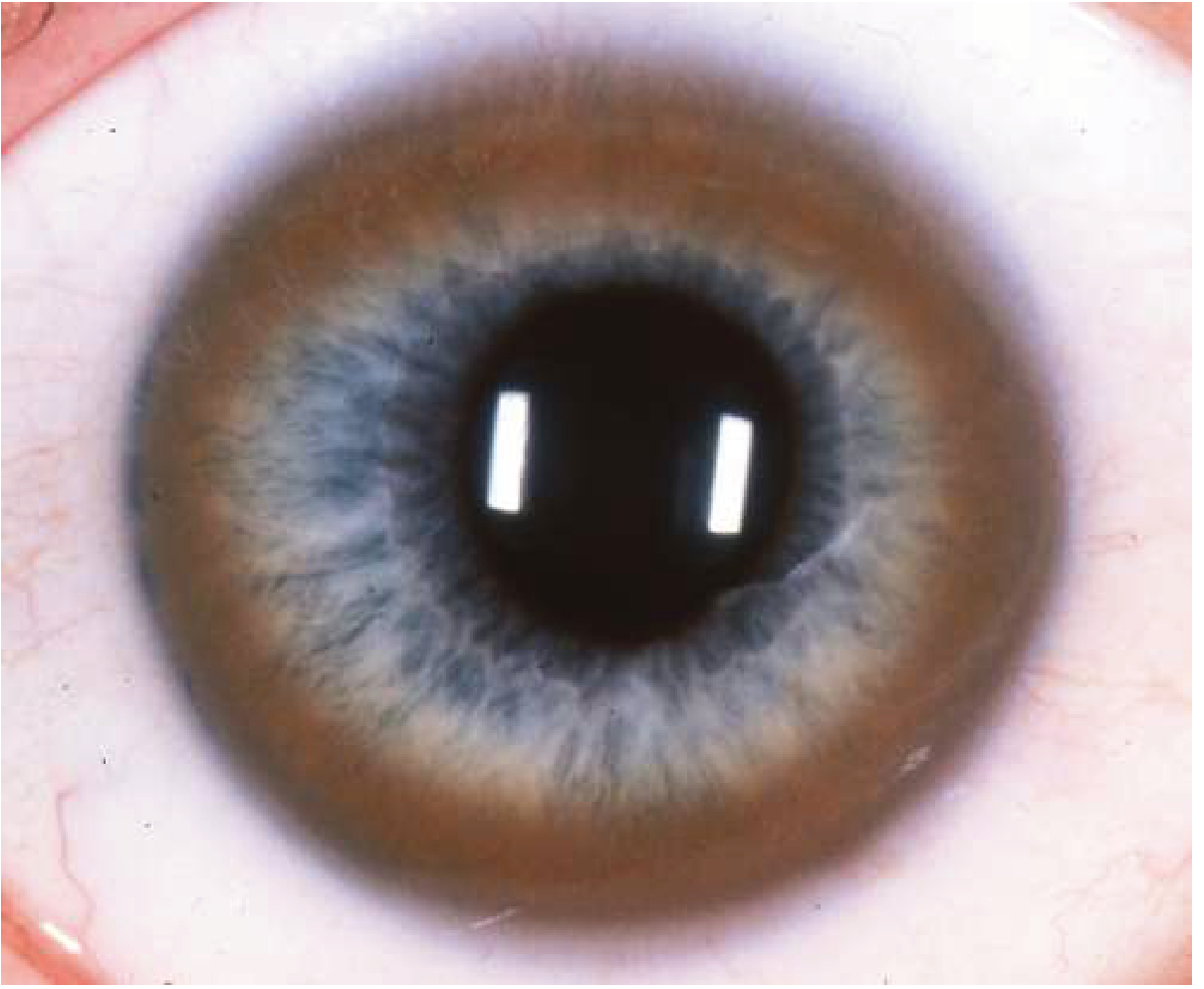
- Copper deposits in Descemet's membrane at the corneal limbus
- Appear golden-brown to greenish (visible in panel C of the slit-lamp image above as a brownish ring at the iris periphery)
- First appear as a superior crescent, then inferior, then circumferential
- Present in ~95-98% of patients with neurologic disease and 50-65% of those with purely hepatic presentation
- Require slit-lamp examination to detect in early stages; may be visible to naked eye when prominent
- Also: "Sunflower" cataracts from copper deposition in the lens (anterior capsular pattern)
- KF rings fade and disappear with effective treatment
5. Other Organ Involvement
| System | Manifestation | Mechanism |
|---|---|---|
| Hematologic | Coombs-negative hemolytic anemia | Direct copper toxicity to RBC membranes |
| Renal | Fanconi syndrome (aminoaciduria, phosphaturia, glycosuria, uricosuria) | Proximal tubular copper toxicity |
| Skeletal | Osteoporosis, rickets, premature osteoarthritis (knees, wrists) | Renal Ca/PO4 losses + direct copper deposition in bone/cartilage |
| Endocrine | Delayed puberty, amenorrhea | Secondary to liver disease |
| Parathyroid | Hypoparathyroidism | Copper deposition |
Diagnosis
Wilson disease is suspected in:
- Unexplained liver disease (especially in young patients)
- ALF with hemolysis + low alkaline phosphatase
- Young adults with neurologic or psychiatric disease + abnormal LFTs
- First-degree relatives of a known patient
Laboratory Findings
| Test | Finding in Wilson Disease | Notes |
|---|---|---|
| Serum ceruloplasmin | <11.5 mg/dL (normal 20-40 mg/dL) | Most efficient first test; can be normal in early disease |
| 24-hour urine copper | >100 μg/day (normal <40 μg/day) | Most specific test; >40 μg/day is suspicious |
| Serum copper | Usually low (but can be low, normal, or elevated) | Not reliable diagnostically |
| Hepatic copper quantification (liver biopsy) | >250 μg/g dry weight (normal 20-50 μg/g) | Most sensitive (~80%); used when other tests inconclusive |
| Liver enzymes | Elevated AST/ALT | |
| Coagulation | Elevated INR | Hepatic dysfunction |
| Coombs test | Negative hemolysis | Important clue in ALF |
| Uric acid | Low (uricosuria) | Fanconi syndrome |
Genetic Testing
- Two disease-specific ATP7B mutations (one on each allele) is diagnostic
- Over 500 mutations complicate sequencing - used mainly in family screening once a proband's mutations are known
Leipzig Scoring System
The Leipzig score (used in clinical practice) combines KF rings, neuropsychiatric symptoms, Coombs-negative hemolysis, serum ceruloplasmin, urine copper, liver copper, and genetic findings. Score ≥4 = Wilson disease; Score 3 = probable; ≤1 = unlikely.
Pathology (Morphology)
Hepatic changes are variable and mimic many other conditions:
- Early: Steatosis (fatty change), focal hepatocyte necrosis
- Intermediate: Chronic hepatitis pattern with inflammation, Mallory hyaline bodies, ballooned hepatocytes (resembles steatohepatitis)
- Advanced: Macronodular cirrhosis
- Special stains: Rubeanic acid or rhodanine stain shows granular copper deposits in hepatocytes (lysosomal distribution)
Brain pathology: Copper deposition primarily in basal ganglia (putamen most prominently), with neuronal loss and gliosis.
Treatment
Treatment is lifelong. Stopping treatment leads to symptomatic relapse and potentially fatal liver failure.
1. Copper Chelation (Symptomatic Patients, Initial Therapy)
D-Penicillamine
- Mechanism: Binds copper → markedly increases urinary copper excretion
- Dose: Start 250 mg twice daily, titrate to 15-20 mg/kg/day in 2-4 divided doses
- Always co-prescribe pyridoxine (vitamin B6) to prevent B6 deficiency
- Side effects: Hypersensitivity, nephrotoxicity (membranous nephropathy), hematologic abnormalities, elastosis perforans serpiginosa (skin rash at neck/axilla)
- Major concern: Paradoxical neurological worsening in 20-50% of patients with neurologic presentations - can be permanent
Trientine (triethylene tetramine dihydrochloride)
- Preferred over penicillamine when neurological disease is the presentation
- Fewer side effects; noninferior for maintenance therapy
- Lower risk of neurological deterioration
- Also used in penicillamine-intolerant patients
Tetrathiomolybdate (ammonium tetrathiomolybdate)
- Forms tripartite complexes with albumin and copper → biliary excretion
- Fast-acting: restores copper balance within weeks vs. months for other agents
- Especially suited for initial treatment of neurologic Wilson disease (least neurological deterioration risk)
- Still investigational in some settings
2. Zinc Therapy (Presymptomatic, Maintenance, Pregnancy)
- Mechanism: Induces intestinal metallothionein → blocks copper absorption from the gut; also promotes fecal copper excretion
- Takes 4-6 months to restore copper balance as monotherapy
- Preferred in presymptomatic patients, pregnant women (safer fetal profile than chelators), and maintenance after initial chelation
- Side effects: Dyspepsia (10-20%); slight higher rate of hepatic decompensation with long-term monotherapy vs. chelation
3. Dietary Restriction
- Avoid copper-rich foods: shellfish, liver/organ meats, mushrooms, chocolate, nuts
- Test drinking water for copper; avoid if >1.3 mg/L
- Restriction alone is insufficient - always combine with pharmacotherapy
4. Liver Transplantation
- Curative - corrects the underlying metabolic defect (functioning hepatocytes carry normal ATP7B)
- Indicated for: Acute liver failure unresponsive to medical therapy; end-stage irreversible cirrhosis
- Long-term outcomes are excellent; disease does not recur in the graft
- A 2025 systematic review and meta-analysis confirms excellent post-transplant outcomes in Wilson disease patients
- Neurological/psychiatric disease without liver failure is generally NOT an indication for transplant
5. Supportive Therapy
- Speech therapy, physical therapy, occupational therapy for neurological patients
- Treatment of tremor/dystonia
- Psychological support
- Genetic counseling and family screening of all first-degree relatives
6. Emerging Therapies
- AAV gene therapy (adeno-associated virus delivering ATP7B): Phase I/II trials ongoing; potentially curative without transplantation - Goldman-Cecil Medicine
- Deep brain stimulation (DBS): A 2025 multicenter meta-analysis shows benefit for severe refractory dystonia in Wilson disease
Summary of Drug Choices by Clinical Scenario
| Scenario | First Choice | Alternative |
|---|---|---|
| Symptomatic liver disease | D-penicillamine or trientine | - |
| Neurologic presentation | Trientine or tetrathiomolybdate | Avoid D-penicillamine if possible |
| Presymptomatic | Zinc | Trientine |
| Maintenance after chelation | Zinc or trientine | - |
| Pregnancy | Zinc | - |
| ALF / end-stage liver disease | Liver transplantation | - |
Prognosis
- With treatment: Generally favorable. Most neurological, psychiatric, renal, and hepatic manifestations stabilize or improve. KF rings fade with effective therapy.
- Without treatment: Progressive and fatal (liver failure, neurological deterioration)
- Non-compliance: Inevitable recurrence; long-term mortality ~3-fold higher than the general population
- Patients who develop acute liver failure have high short-term mortality without transplantation
Sources: Goldman-Cecil Medicine | Robbins & Kumar Basic Pathology | Bradley & Daroff's Neurology in Clinical Practice | Sleisenger & Fordtran's GI and Liver Disease | Kanski's Clinical Ophthalmology | Current Surgical Therapy
This is a shared conversation. Sign in to Orris to start your own chat.