Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathogenesis
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism alveolar capillary damage
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by diffuse alveolar damage (DAD), severe inflammation, and profound hypoxemia. The Berlin Definition requires bilateral infiltrates on imaging, PaO₂/FiO₂ <300 mmHg on ≥5 cmH₂O PEEP, onset within 7 days of a known insult, and exclusion of cardiogenic edema.
1. Initiating Insults — Direct vs. Indirect Pathways
Lung injury in ARDS arrives via two broad routes that ultimately converge:
| Route | Examples |
|---|---|
| Direct (pulmonary) | Pneumonia, aspiration, inhalation injury, near-drowning |
| Indirect (extrapulmonary) | Sepsis, severe pancreatitis, trauma, massive transfusion (TRALI), shock |
Extrapulmonary sepsis is the most common cause (ARDS develops in 20–45% of patients with severe sepsis). Injury via the bloodstream requires intermediary mechanisms — chiefly the activated neutrophil — whereas direct insults act by contact with lung tissue. These pathways overlap, since once injury begins, inflammatory amplification takes over regardless of origin. — Fishman's Pulmonary Diseases and Disorders, p. 2482
2. The Alveolar-Capillary Barrier — The Central Target
The alveolar-capillary membrane consists of:
- Type I pneumocytes (95% of surface area, thin gas exchange cells — highly vulnerable)
- Type II pneumocytes (surfactant producers, progenitor cells for repair)
- Pulmonary capillary endothelium
- Shared basement membrane
In ARDS, both the epithelial and endothelial layers are injured, leading to loss of their tight junctions and increased permeability. The result is flooding of the alveolar space with protein-rich edema fluid, cellular debris, and neutrophils.
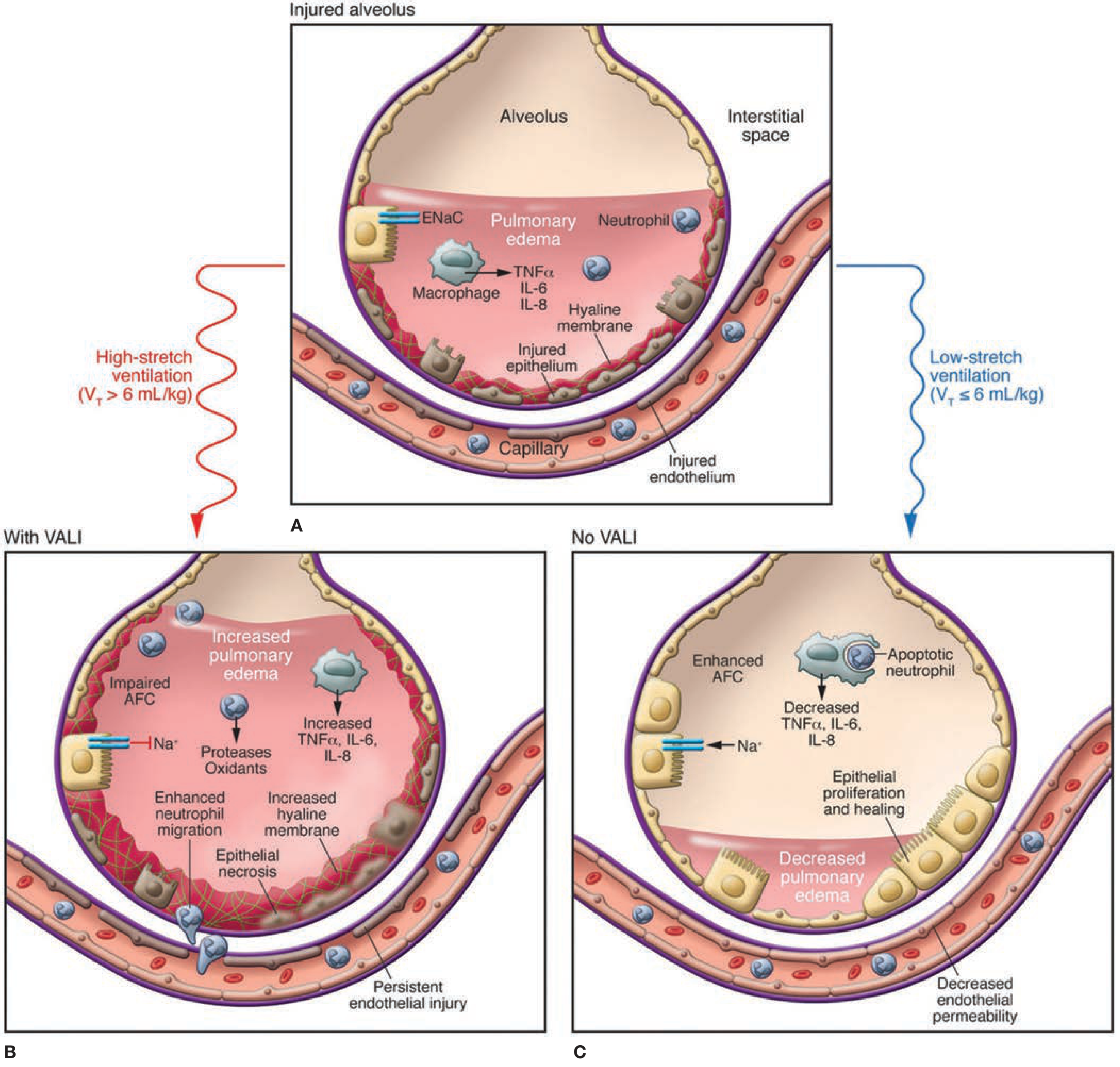
Panel A shows the injured alveolus: pulmonary edema filling the air space, activated macrophages releasing TNF-α/IL-6/IL-8, neutrophils migrating in, hyaline membrane formation, and injury to both epithelium and endothelium.
3. The Inflammatory Cascade
This is the core of ARDS pathogenesis and involves multiple overlapping mediator systems:
A. Macrophage Activation & Cytokine Storm
- Alveolar macrophages recognize pathogen-associated molecular patterns (PAMPs, e.g., LPS) and damage-associated molecular patterns (DAMPs) via Toll-like receptors (TLRs)
- Activated macrophages release TNF-α, IL-1β, IL-6, IL-8 (CXCL8), and other pro-inflammatory cytokines
- These act as chemoattractants, pulling circulating neutrophils into the lung
B. Neutrophil Accumulation and Activation
Neutrophils are central effectors of ARDS injury. Their pathologic sequence:
- Margination — Neutrophils adhere to injured endothelium via upregulated adhesion molecules (ICAM-1, selectins)
- Transmigration — They cross the endothelium and epithelium into the alveolar space, driven by IL-8 and other chemokines
- Activation — Release destructive mediators including:
- Reactive oxygen species (ROS) — oxidative damage to membranes and proteins
- Proteases (elastase, MMP-8, MMP-9) — degrade basement membrane and tight junctions
- Myeloperoxidase — generates hypochlorous acid (HOCl)
- Platelet-activating factor (PAF) and leukotrienes — amplify vascular permeability
BAL studies in patients show the inflammatory response begins before clinical ARDS recognition, peaks at days 1–3, and resolves over 7–14 days. — Fishman's, p. 2481
C. Lipid Mediators
- Phospholipase A2 (especially in pancreatitis) degrades surfactant phospholipids and directly increases vascular permeability
- Leukotriene B4 (LTB4) is a potent neutrophil chemoattractant
- PAF promotes platelet aggregation and neutrophil activation
D. Coagulation-Inflammation Cross-talk
- Plasma proteins leaking into alveoli activate the coagulation cascade
- Fibrin deposition forms hyaline membranes (the histologic hallmark of DAD)
- Tissue factor is upregulated on macrophages and endothelium
- Simultaneous inhibition of fibrinolysis (↑PAI-1) traps fibrin in alveoli
- Platelet aggregation within microvessels contributes to microvascular thrombosis and dead-space ventilation
E. Angiopoietins
- Angiopoietin-2 (Ang-2) is released from activated endothelium (Weibel-Palade bodies), competitively inhibiting Tie-2 receptor signaling and destabilizing endothelial junctions
- Elevated Ang-2 is a biomarker of endothelial activation in ARDS
- Ang-1 (from pericytes) opposes this and promotes barrier stability
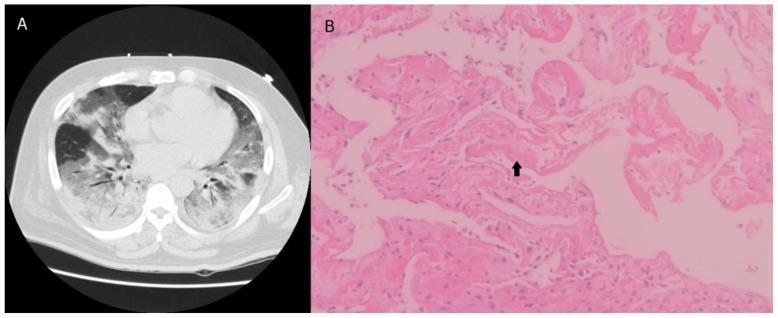
4. Diffuse Alveolar Damage — The Pathological Result
DAD progresses in two phases:
| Phase | Timing | Histology |
|---|---|---|
| Exudative | Days 0–7 | Alveolar edema, hyaline membranes, type I cell necrosis, neutrophil infiltration, microvascular fibrin thrombi |
| Proliferative/Organizing | Days 7–21+ | Type II cell hyperplasia (attempted repair), fibroblast proliferation, organizing fibrosis, residual inflammation |
5. Surfactant Dysfunction
- ARDS dramatically impairs surfactant function through multiple mechanisms:
- Type II pneumocyte injury → reduced surfactant production (phosphatidylcholine, SP-B, SP-C)
- Phospholipase A2 → enzymatic degradation of existing surfactant
- Plasma protein leak (albumin, fibrin) into alveoli → inactivation of surfactant function
- Loss of surfactant raises alveolar surface tension → alveolar collapse (atelectasis) → reduced functional residual capacity (FRC) → severe hypoxemia
6. Sodium/Water Transport Failure
Normal alveolar fluid clearance (AFC) depends on vectorial Na⁺ transport via epithelial sodium channels (ENaC) on type I and II cells, creating an osmotic gradient to reabsorb edema. In ARDS:
- Apoptosis and necrosis of alveolar epithelium destroy this machinery
- Proinflammatory cytokines, oxidants, and hypoxia impair transcellular ion transport
- AFC rate falls dramatically → fluid accumulates and worsens gas exchange — Fishman's, p. 2484
7. Ventilator-Induced Lung Injury (VILI) — A Compounding Mechanism
Once ARDS is established, mechanical ventilation can perpetuate and worsen injury:
- Volutrauma/barotrauma — overdistension of open lung units at high tidal volumes
- Atelectrauma — repetitive collapse and reopening of dependent lung units
- Both mechanisms trigger biotrauma — cytokine release (TNF-α, IL-6, IL-8) that can spill into the systemic circulation, driving multi-organ failure
This is why low tidal volume ventilation (6 mL/kg IBW, plateau pressure ≤30 cmH₂O) is the only ventilatory strategy with proven mortality benefit. — Fishman's VALI diagram, p. 2485; Sabiston, p. 940
Summary Diagram of Pathogenesis
Direct insult (pneumonia, aspiration)
↓
Indirect insult (sepsis, pancreatitis)
↓
Alveolar macrophage activation (TLR → TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil margination, transmigration, activation
↓
ROS + proteases → tight junction disruption
↓
Alveolar-capillary barrier breach → protein-rich edema floods alveoli
↓
Type I cell necrosis + Type II cell injury → surfactant failure
↓
ENaC dysfunction → impaired fluid clearance
↓
Coagulation activation → fibrin/hyaline membranes
↓
Diffuse alveolar damage: bilateral opacities, severe hypoxemia, ↓compliance
↓ ↑
VILI (if high tidal volume)
Key Biomarkers Reflecting These Mechanisms
| Biomarker | Source | What it reflects |
|---|---|---|
| IL-8 | Macrophages, epithelium | Neutrophil chemotaxis |
| IL-6 | Macrophages | Systemic inflammation |
| Ang-2 | Endothelium | Endothelial activation |
| SP-D | Type II cells | Epithelial injury |
| RAGE | Type I cells | Alveolar epithelial necrosis |
| PAI-1 | Endothelium/macrophages | Fibrinolysis inhibition |
Sources:
- Fishman's Pulmonary Diseases and Disorders — Mechanisms of ARDS chapter, pp. 2480–2485
- Murray & Nadel's Textbook of Respiratory Medicine — Etiology and Pathogenesis of ARDS chapter
- Sabiston Textbook of Surgery — ARDS, p. 940
- Goldman-Cecil Medicine — Chapter 90/91
This is a shared conversation. Sign in to Orris to start your own chat.