Hematuria Proteinuria Polyuria Dialysis Hemodialysis Renal Osteodystrophy Microalbuminuria Diabetic Kidney Disease Acute Tubular Necrosis Drug-induced Renal Disease
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Nephrology: Core Concepts
1. Hematuria
Hematuria is the presence of red blood cells in the urine. It is classified as:
- Gross hematuria: visible blood, apparent to patient or clinician
- Microscopic hematuria: defined as ≥3 RBCs/high-power field (hpf) in adults; in children, >5 RBCs/hpf on at least two weekly samples is considered abnormal
One prospective study found 61% of patients with hematuria had no significant finding after evaluation. However, the malignancy rate in screening studies is ~2.6%, justifying workup. Most patients with hematuria do not have serious pathology, but this depends on type and risk factors.
Causes include:
- Urinary tract infection (cystitis, pyelonephritis)
- Kidney stones (nephrolithiasis)
- Glomerulonephritis (RBC casts indicate glomerular origin)
- Bladder or renal malignancy
- Benign prostatic hyperplasia
- Trauma, vigorous exercise
- Medications (e.g., anticoagulants, cyclophosphamide)
- IgA nephropathy (episodic gross hematuria post-infection)
Evaluation depends on whether gross or microscopic, and patient age. Adults with microscopic hematuria require UPr/UCr measurement and evaluation for CKD or urological malignancy.
- Textbook of Family Medicine 9e; Brenner and Rector's The Kidney; Campbell Walsh Wein Urology
2. Proteinuria
The kidneys normally excrete only small amounts of protein (mainly glycoproteins) daily. Consistent albuminuria implies glomerular or tubular dysfunction.
Classification by 24-hour urine:
| Category | Value |
|---|---|
| Normal | <30 mg/24 hr |
| Microalbuminuria | 30-300 mg/24 hr |
| Macroalbuminuria (clinical proteinuria) | >300 mg/24 hr |
| Nephrotic-range proteinuria | >3.5 g/24 hr |
Measurement:
- Urine dipstick: convenient but less precise
- Urine protein/creatinine (UPr/UCr) ratio: best random-sample test; correlates well with 24-hour excretion; normal adult value <0.2
- 24-hour urine collections are generally not needed
Mechanisms of proteinuria:
- Glomerular injury: podocyte foot process effacement, disruption of the slit pore membrane protein nephrin - the key structural element of the filtration barrier
- Angiotensin II dramatically decreases nephrin expression in podocytes; ACE inhibitors restore it
- Tubular dysfunction: failure to reabsorb filtered proteins
Types in children:
- Functional (fever, exercise) - transient
- Orthostatic (benign) - isolated
- Persistent proteinuria warrants evaluation for CKD; found on ≥2 of 3 weekly samples
Clinical significance: Proteinuria is not just a marker of kidney disease - it is a pathogenetic factor in the progression of renal dysfunction.
- Textbook of Family Medicine 9e; Brenner and Rector's The Kidney
3. Polyuria
Definition: Urine volume >2.5 L/day (conventional); a physiology-based definition considers polyuria present when urine flow is higher than expected for the rate of osmole excretion in a specific setting.
Two categories:
A. Water Diuresis
Caused by failure of vasopressin (ADH) action or production:
- Central diabetes insipidus - insufficient ADH release
- Nephrogenic diabetes insipidus - renal resistance to ADH
- Primary polydipsia - excess water intake suppresses ADH
Key physiology: Water reabsorption depends on aquaporin (AQP) water channels:
- AQP1: constitutively present in proximal tubule and descending thin limb (non-regulated)
- AQP2: in collecting duct principal cells - regulated by vasopressin; vasopressin causes AQP2 insertion into luminal membrane
Without vasopressin action, urine flow can reach 10-15 mL/min (~14-22 L/day).
B. Osmotic Diuresis
Caused by high concentrations of effective osmoles in the tubular fluid:
-
Diabetes mellitus (glucosuria)
-
Mannitol administration
-
High protein/urea load
-
Post-obstructive diuresis
-
Brenner and Rector's The Kidney; Goldman-Cecil Medicine
4. Dialysis
Dialysis is renal replacement therapy (RRT) used when the kidneys can no longer maintain fluid, electrolyte, and waste homeostasis. It is indicated in end-stage kidney disease (ESKD) or severe acute kidney injury.
Two main modalities:
- Hemodialysis (HD)
- Peritoneal Dialysis (PD)
Choosing between HD and PD:
There is a notable absence of comparative studies proving superiority of one modality over another. The choice is based on:
- Center experience
- Patient and family preference
- Socioeconomic circumstances and treatment availability
- Compliance capability
- Clinical factors (vascular access, peritoneal membrane integrity)
PD advantages:
- More liberal fluid intake (important in infants fed liquid nutrition)
- Can be done at home
- Better school attendance in pediatric patients (74-85% vs 47-59% full schooling)
- Often the only option in areas without HD centers
HD considerations:
-
Requires travel to dialysis center at least 3x/week
-
In-center HD is time-consuming and costly
-
In the US, trend has shifted from 34% HD in 1991 to 54% HD in 2010
-
Brenner and Rector's The Kidney; Comprehensive Clinical Nephrology, 7th Ed.
5. Hemodialysis
Hemodialysis (HD) uses an extracorporeal circuit to filter blood through a semipermeable membrane (dialyzer), removing uremic toxins, excess fluid, and electrolytes.
Vascular Access - three main types:
| Access Type | Notes |
|---|---|
| AV Fistula | Preferred; lowest complication rate; median survival ~3.14 years |
| AV Graft | Synthetic; higher complication rate than fistula |
| Central Venous Catheter (CVC) | For short-term or emergent use; median survival ~0.6 year; highest infection and malfunction risk |
AV fistula is the recommended choice because it has:
- Lower rate of infection and malfunction
- Longer access longevity
- Better clearance and higher albumin/hemoglobin levels
CVC limitations: Central venous stenosis from CVC use may preclude future ipsilateral AV fistula creation - especially important in younger patients expected to require long-term RRT.
Indications for hemodialysis initiation (in AKI or CKD):
- Severe hyperkalemia refractory to medical management
- Metabolic acidosis unresponsive to treatment
- Pulmonary edema/fluid overload
- Uremic encephalopathy, pericarditis, or bleeding
- Certain drug/toxin ingestions (e.g., methanol, ethylene glycol, lithium)
Adequacy: Measured by Kt/V (K = dialyzer clearance, t = dialysis time, V = volume of distribution of urea). Target Kt/V ≥1.2 per session for thrice-weekly HD.
- Brenner and Rector's The Kidney; Comprehensive Clinical Nephrology, 7th Ed.
6. Renal Osteodystrophy
Renal osteodystrophy (ROD) is the spectrum of bone abnormalities that occur in patients with end-stage renal disease (ESRD).
Pathogenesis:
- Metabolic disturbances: hypocalcemia, hyperphosphatemia
- Hormonal disturbances: secondary hyperparathyroidism (2° HPT)
- Decreased renal synthesis of 1,25(OH)₂D₃ (calcitriol) → decreased intestinal calcium absorption → hypocalcemia → PTH elevation
- Phosphate retention (reduced GFR) further suppresses calcitriol and directly stimulates PTH
Two main histologic patterns:
Osteitis Fibrosa Cystica
- Due to secondary hyperparathyroidism
- Increased bone turnover: accelerated bone dissolution, decreased bone formation
- PTH drives osteoclast activity
- Brown tumors (fibrous cysts) may form
Osteomalacia
- Poor mineralization of bone matrix (osteoid accumulation)
- Osteoclasts cannot resorb unmineralized osteoid surfaces - only mineralized surfaces
- Osteoclasts must dig "tunneling resorption" through remaining mineralized areas
- Historically linked to aluminum toxicity from phosphate binders (aluminum incorporated into hydroxyapatite, blocking further mineralization)
Histologic appearance (Von Kossa stain):
- Mineralized bone stains black
- Osteoid (unmineralized) stains magenta/red
- Thick osteoid seams = osteomalacia
- Scalloped osteoclastic resorption = osteitis fibrosa
Patients with ESRD have increased fracture risk - predominantly osteitis fibrosa cystica, osteomalacia, or a combination of both.
Management:
- Phosphate binders (calcium-based or non-calcium: sevelamer, lanthanum)
- Active vitamin D analogues (calcitriol, paricalcitol)
- Calcimimetics (cinacalcet) to suppress PTH
- Dietary phosphate restriction
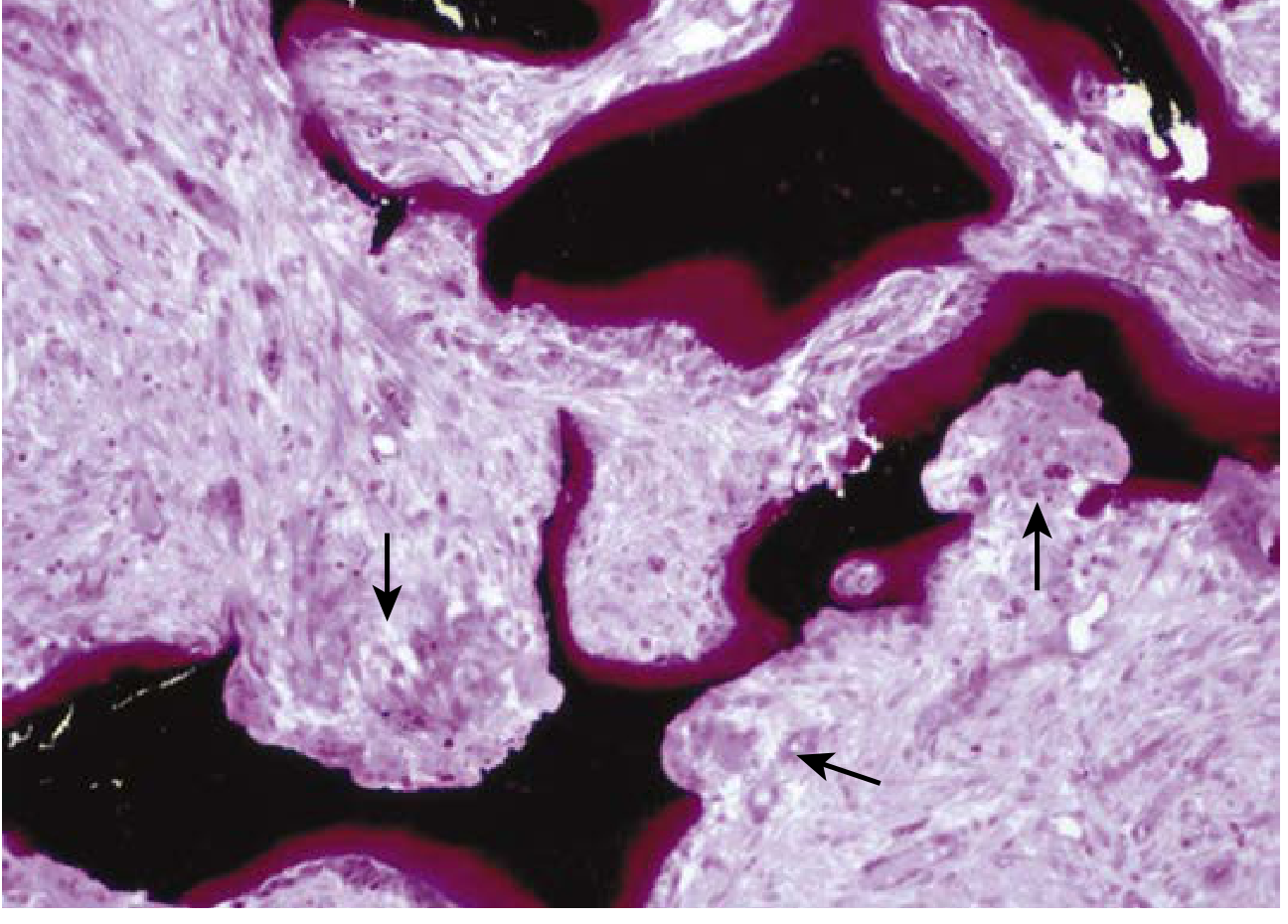
Renal osteodystrophy: Von Kossa stain showing thick osteoid seams (red/magenta) and osteoclastic tunneling resorption of mineralized bone (black). Arrows indicate cutting cones of osteoclasts. - Henry's Clinical Diagnosis and Management by Laboratory Methods
- Henry's Clinical Diagnosis; Robbins Pathology; Grainger & Allison's Diagnostic Radiology
7. Microalbuminuria
Microalbuminuria refers to moderately increased albuminuria: albumin excretion of 30-300 mg/24 hours (or 30-300 mg/g creatinine on spot urine).
Significance:
- It is below the detection threshold of standard urine dipstick (which detects protein only when >300 mg/day)
- It is a marker of early glomerular damage
- A powerful predictor of progressive nephropathy in diabetic patients
- Also strongly predicts cardiovascular mortality
In diabetic patients:
- Approximately 30% of type 1 diabetics progress to microalbuminuria (stage 2 diabetic nephropathy) after ~10 years
- Microalbuminuria and macroalbuminuria significantly increase mortality risk: prospective data from 328 patients with type 2 diabetes showed macroalbuminuria and microalbuminuria both led to markedly elevated 5-year mortality
- RAAS blockers (ACE inhibitors, ARBs) can delay onset of microalbuminuria: the ROADMAP trial showed olmesartan produced a 23% delay in time to onset of microalbuminuria in hypertensive patients with type 2 diabetes
Screening: Patients with diabetes should be periodically tested for microalbuminuria. Those with hypertension and other CKD risk factors should also be tested.
Why dipstick misses it: Standard dipstick detects albumin ≥300 mg/day; albumin-specific dipstick or laboratory measurement (ACR) required for microalbuminuria detection.
- Goldman-Cecil Medicine; Brenner and Rector's The Kidney; Textbook of Family Medicine 9e
8. Diabetic Kidney Disease (Diabetic Nephropathy)
Diabetic kidney disease (DKD) is the leading cause of ESKD in many countries. In Japan, it overtook glomerulonephritis as the top cause of ESKD in dialysis patients since 1997.
Prevalence: ~30-40% of type 2 diabetic patients develop diabetic nephropathy.
Staging (classical Mogensen stages for type 1):
| Stage | Features |
|---|---|
| I - Hyperfiltration | Elevated GFR, kidney enlargement |
| II - Silent | Glomerular lesions, normoalbuminuria or microalbuminuria |
| III - Incipient nephropathy | Fixed microalbuminuria (30-300 mg/day) |
| IV - Overt nephropathy | Macroalbuminuria (>300 mg/day), declining GFR |
| V - ESKD | GFR <15, requires renal replacement therapy |
Pathogenesis - key mechanisms:
- Hemodynamic: Hyperglycemia → afferent arteriolar dilation → increased glomerular capillary pressure (PGC) → hyperfiltration → glomerular hypertrophy
- RAAS activation: Despite low plasma renin (paradoxically), prorenin is elevated; the RAS plays a key role in progressive injury
- Podocyte injury: Angiotensin II decreases nephrin expression; ACE inhibitor treatment restores nephrin levels
- Genetic factors: ACE gene I/D polymorphism - DD genotype associated with higher ACE activity and greater susceptibility to ESKD; IL-10, IL-6, ICAM-1 polymorphisms also implicated
- Inflammation: Angiotensin II activates NF-κB → upregulates MCP-1, RANTES, IL-6 → macrophage infiltration
The RAAS paradox in DKD: Despite characteristically low plasma renin in long-standing diabetes, RAAS blockers are the mainstay of therapy - explained partly by elevated prorenin and local (intrarenal) RAAS activation.
Management:
-
Glycemic control (HbA1c target ~7%)
-
ACE inhibitors or ARBs: first-line antiproteinuric and renoprotective therapy
-
SGLT-2 inhibitors (e.g., empagliflozin, dapagliflozin): proven to slow CKD progression
-
Blood pressure control (<130/80 mmHg)
-
Dietary protein restriction
-
Brenner and Rector's The Kidney; Goldman-Cecil Medicine
9. Acute Tubular Necrosis (ATN)
ATN is a clinical syndrome of abrupt, sustained decline in GFR triggered by an acute ischemic or nephrotoxic event, developing within minutes to days after the insult.
Epidemiology: ATN is the most common cause of AKI in hospitalized patients (just under half of all cases); in the ICU, it accounts for more than half of AKI cases.
Causes:
| Category | Examples |
|---|---|
| Ischemic/prerenal spectrum | Cardiogenic shock, septic shock, severe volume depletion, major surgery |
| Nephrotoxic - exogenous | Aminoglycosides, amphotericin B, cisplatin, radiocontrast agents, NSAIDs |
| Nephrotoxic - endogenous | Myoglobin (rhabdomyolysis), hemoglobin (hemolysis), uric acid (tumor lysis), myeloma light chains |
Pathophysiology:
- Obstruction of tubular flow by cellular debris
- Disruption of tubular cell polarity and cytoskeleton
- Backleak of glomerular filtrate through denuded epithelium into interstitium
- Afferent arteriolar vasoconstriction
- Loss of microvasculature and immune cell infiltration
ATN has been described as a "(mal)adaptive response" - trading away GFR to preserve medullary oxygenation and tubular integrity. Ischemic ATN is most severe in the outer medulla (proximal straight tubule and thick ascending limb are most vulnerable).
Histology: Vacuolation, loss of brush border, disruption of epithelial lining, intratubular casts. Necrosis is focal and may be missed on biopsy (which samples cortex, not outer medulla well). Regenerative changes and fresh injury often coexist.
Diagnosis:
| Finding | Significance |
|---|---|
| FENa >1-2% | Suggests ATN (intrinsic renal) vs prerenal (<1%) |
| Fractional excretion of urea >35-50% | Used when diuretics confound FENa |
| "Muddy brown" granular casts | Hallmark of ATN on urine microscopy; ATN highly likely with ≥6 granular casts |
| Rising BUN and creatinine | Within days of insult |
Clinical course:
- Initiation phase (hours to days)
- Maintenance/oliguric phase (1-2 weeks typically)
- Recovery/diuretic phase

"Muddy brown" granular cast - the hallmark urinary finding of acute tubular necrosis. - Frameworks for Internal Medicine
- Frameworks for Internal Medicine; Comprehensive Clinical Nephrology, 7th Ed.; ROSEN's Emergency Medicine
10. Drug-Induced Renal Disease
Drugs can injure the kidney through multiple mechanisms. Major patterns include:
A. Drug-Induced Acute Tubular Necrosis (Nephrotoxicity)
Direct tubular epithelial toxicity:
- Aminoglycosides: proximal tubular toxicity (accumulate in lysosomes)
- Amphotericin B: direct tubular toxicity + afferent arteriolar constriction
- Cisplatin: mitochondrial injury and DNA damage in proximal tubule
- Contrast nephropathy: ischemic + toxic mechanisms; risk increases with CKD, diabetes, volume depletion
B. Drug-Induced Acute Interstitial Nephritis (AIN)
Most common cause of AIN; drug use accounts for the majority of cases (particularly antimicrobials, proton pump inhibitors, NSAIDs):
- Mean latency from drug exposure to kidney manifestations: ~10 days (can be 1 day to months)
- Classic triad: fever (36%), rash (22%), arthralgias (45%) - but often incomplete
- Urinary findings: nonnephrotic proteinuria (93%), leukocyturia (82%), microscopic hematuria (67%)
- Diagnosis usually requires kidney biopsy
C. Drug-Induced Tubular Secretory Defects (Electrolyte disturbances)
| Drug | Mechanism | Effect |
|---|---|---|
| NSAIDs/COX-2 inhibitors | Inhibit renin release | Hyperkalemia, metabolic acidosis |
| ACE inhibitors/ARBs | Block renin-angiotensin | Hypoaldosteronism → hyperkalemia |
| Heparin | Damages zona glomerulosa; inhibits aldosterone synthase | Hyperkalemia |
| Spironolactone/eplerenone | Competitive aldosterone inhibitors | Hyperkalemia (especially in renal insufficiency) |
| Amiloride/triamterene | Block ENaC in collecting duct | Hyperkalemia, reduced K⁺ secretion |
| Trimethoprim/pentamidine | Structurally similar to amiloride; block ENaC | Hyperkalemia in 20-50% (TMP-SMX); metabolic acidosis |
| Cyclosporine A/tacrolimus | Inhibit Na⁺-K⁺-ATPase; block ROMK K⁺ channel | Hyperkalemia in transplant recipients |
Important note: The combination of potassium-sparing diuretics + ACE inhibitors should be avoided in diabetic patients (who already have hyporeninemic hypoaldosteronism tendency).
D. Lithium Nephrotoxicity
- Chronic lithium use causes nephrogenic diabetes insipidus and chronic tubulointerstitial nephritis
- Can progress to CKD
E. Calcineurin Inhibitor Nephrotoxicity
-
Cyclosporine/tacrolimus: cause both acute (afferent arteriolar vasoconstriction) and chronic (fibrosis/striped interstitial fibrosis) nephrotoxicity
-
Brenner and Rector's The Kidney; Comprehensive Clinical Nephrology, 7th Ed.; Goodman & Gilman's Pharmacology
Quick Reference Summary
| Topic | Key Point |
|---|---|
| Hematuria | ≥3 RBCs/hpf (adults); 2.6% malignancy rate in screening |
| Proteinuria | UPr/UCr ratio best test; >3.5 g/day = nephrotic range |
| Polyuria | >2.5 L/day; water diuresis (ADH/AQP2) vs osmotic diuresis |
| Dialysis | HD vs PD - no proven superiority; choice based on patient factors |
| Hemodialysis | AV fistula preferred access; target Kt/V ≥1.2 |
| Renal Osteodystrophy | Secondary HPT (osteitis fibrosa) + poor mineralization (osteomalacia) |
| Microalbuminuria | 30-300 mg/day; below dipstick threshold; early DKD marker |
| Diabetic Kidney Disease | Leading cause of ESKD; RAAS blockers + SGLT-2i are cornerstones |
| ATN | Most common hospital AKI; "muddy brown" casts; FENa >1-2% |
| Drug-Induced Renal Disease | ATN (aminoglycosides, contrast), AIN (NSAIDs, PPIs), electrolyte defects (TMP, heparin) |
This is a shared conversation. Sign in to Orris to start your own chat.