Pathophysiology of myasthenia gravis.. how does pyridostigmine helps
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive, well-cited answer. Let me compose it.
Myasthenia Gravis: Pathophysiology and How Pyridostigmine Helps
1. Normal Neuromuscular Junction (NMJ) - The Baseline
Understanding MG requires first understanding normal NMJ physiology. When a motor nerve fires, it releases acetylcholine (ACh) from synaptic vesicles into the synaptic cleft. ACh binds to nicotinic ACh receptors (AChRs) on the postsynaptic (muscle) membrane, opening ion channels. Cations flow in (Na+ and Ca2+) and K+ flows out, generating an end-plate potential (EPP). Normally, the EPP is enormous - about 70-80 mV relative to the resting potential of -90 mV - far exceeding the -45 mV threshold needed to trigger a muscle action potential. This large buffer above threshold is called the safety factor of neuromuscular transmission. - Eric Kandel, Principles of Neural Science, 6th Ed.
2. Pathophysiology of Myasthenia Gravis
The Core Defect: Postsynaptic Autoimmune Attack
MG is an autoimmune disorder of postsynaptic neuromuscular transmission. The presynaptic nerve terminal is structurally and functionally normal - ACh is synthesized and released in normal amounts. The problem lies entirely on the muscle side. - Bradley and Daroff's Neurology in Clinical Practice
Autoantibodies involved (by frequency):
| Antibody | Frequency | Effect |
|---|---|---|
| Anti-AChR (anti-alpha-1 subunit of nicotinic AChR) | 80-90% of generalized MG; 50% of ocular MG | Receptor destruction, blockade, accelerated degradation |
| Anti-MuSK (muscle-specific tyrosine kinase) | Small % of AChR-negative cases | Disrupts AChR clustering at NMJ; more facial/bulbar involvement |
| Anti-LRP4 (lipoprotein receptor-related protein 4) | 1-3% of all patients | Mild-moderate symptoms |
- Bradley and Daroff's Neurology in Clinical Practice; Tintinalli's Emergency Medicine
Mechanisms by Which Anti-AChR Antibodies Cause Damage
The antibodies harm the NMJ through three mechanisms:
- Receptor blockade - Antibodies physically block the ACh binding site, preventing ACh from attaching to its receptor.
- Accelerated degradation - Antibody binding cross-links adjacent AChRs, triggering endocytosis and lysosomal destruction, causing a net reduction in receptor number.
- Complement-mediated destruction - AChR antibodies are complement-fixing; complement activation destroys the postsynaptic membrane and its junctional folds. The normal infolding of junctional folds is reduced, the synaptic cleft is widened, and synaptic architecture is disrupted.
The result: a 70-90% reduction in the number of functional AChRs per end-plate. - Ganong's Review of Medical Physiology, 26th Ed.; Principles of Neural Science
How This Reduces the Safety Factor and Causes Fatigability
Because so few receptors remain, ACh has a much lower probability of finding a receptor before it is hydrolyzed by acetylcholinesterase (AChE). The EPP amplitude drops to a level that is barely above - or even below - the threshold for a muscle action potential. This narrows or abolishes the safety factor.
At rest, some patients can still activate a muscle action potential just barely. But with repeated stimulation, the normal physiologic decline in ACh quanta released per impulse (normal synaptic fatigue) drops ACh still further, pushing the EPP below threshold in increasing numbers of junctions. Fewer and fewer muscle fibers fire with each successive nerve impulse - producing the characteristic decremental response on repetitive nerve stimulation (RNS) testing, and clinically as progressive fatigability with use.
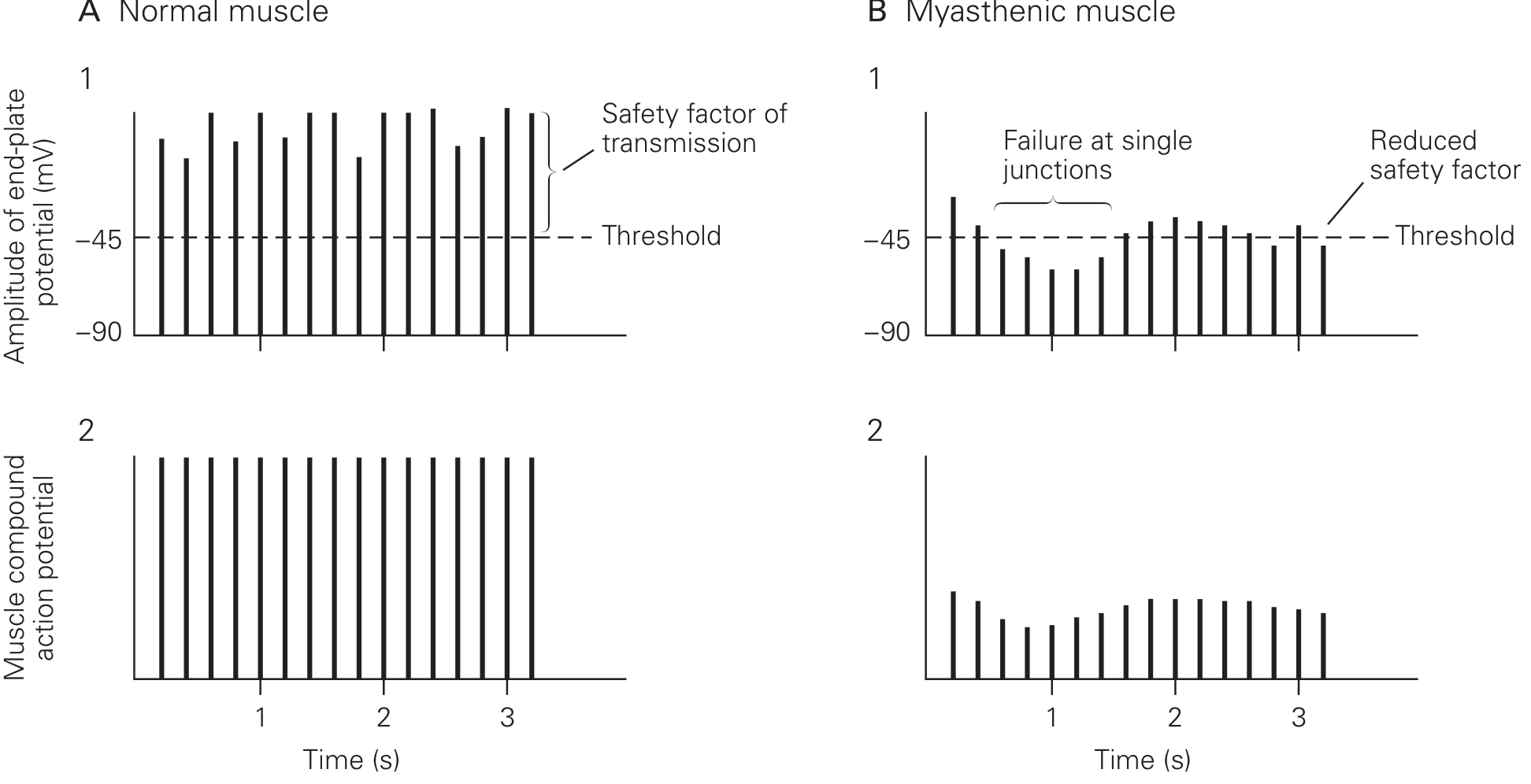
Role of the Thymus
The thymus is abnormal in most MG patients - thymic hyperplasia is present in the majority, and a thymoma in 10-15%. The thymus is thought to supply helper T cells sensitized against thymic proteins that cross-react with AChRs, driving and perpetuating the autoimmune response. This is why thymectomy induces remission in ~35% and improves symptoms in another ~45% of patients. - Ganong's Review; Principles of Neural Science
3. Mechanism of Action of Pyridostigmine
Pyridostigmine is a reversible acetylcholinesterase (AChE) inhibitor - a carbamate ester class drug.
How it works, step by step:
- AChE inhibition - Pyridostigmine binds reversibly to AChE at the NMJ, blocking it from hydrolyzing ACh in the synaptic cleft.
- ACh accumulates - Because ACh is not broken down, its concentration in the cleft rises and it persists for longer.
- Increased receptor occupancy - With more ACh present for longer, the probability that ACh molecules find and bind to the reduced number of remaining receptors increases substantially.
- EPP amplitude restored - More receptor activation generates a larger summed EPP that can more reliably exceed the threshold, restoring muscle action potential generation.
- Safety factor partially restored - Fatigability is reduced because even during sustained activity, enough ACh remains in the cleft to continue activating the available receptors.
In essence: pyridostigmine compensates for having fewer receptors by making each ACh molecule "last longer" and "try harder" to find one. - Costanzo Physiology, 7th Ed.; Lippincott Illustrated Reviews: Pharmacology
4. Pyridostigmine Pharmacology
| Property | Detail |
|---|---|
| Class | Reversible AChE inhibitor (carbamate) |
| Route | Oral (primarily for chronic MG management) |
| Duration of action | 3-6 hours (longer than neostigmine's 30 min-2 hrs) |
| CNS penetration | Quaternary nitrogen = does NOT cross the blood-brain barrier; purely peripheral action |
| Preferred over neostigmine | For chronic outpatient MG due to longer duration and oral bioavailability |
- Lippincott Illustrated Reviews: Pharmacology; Barash Clinical Anesthesia, 9th Ed.
Adverse effects (all from cholinergic excess - muscarinic side effects):
- Salivation, lacrimation, urination, defecation (SLUD)
- Abdominal cramping, diarrhea, nausea
- Bradycardia, bronchospasm
- Miosis
These can be managed with anticholinergic agents (e.g., glycopyrrolate) if needed. No CNS side effects because pyridostigmine does not cross the BBB.
5. Why Pyridostigmine is Symptomatic, Not Curative
Pyridostigmine does not address the underlying autoimmune process. It does not reduce antibody levels or stop AChR destruction. It simply makes better use of the depleted receptors that remain. For disease modification, MG requires immunosuppression (steroids, azathioprine, mycophenolate), plasma exchange or IVIG for rapid improvement, and thymectomy when indicated. Newer agents like eculizumab (anti-C5 complement inhibitor) directly target the complement pathway that destroys the postsynaptic membrane. - Bradley and Daroff's Neurology; Goldman-Cecil Medicine
Key sources:
- Bradley and Daroff's Neurology in Clinical Practice (chapter on NMJ disorders)
- Eric Kandel, Principles of Neural Science, 6th Ed.
- Ganong's Review of Medical Physiology, 26th Ed.
- Costanzo Physiology, 7th Ed.
- Lippincott Illustrated Reviews: Pharmacology
- Tintinalli's Emergency Medicine
Mg vs op poisoning
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Excellent - I have comprehensive content from multiple authoritative textbooks. I have everything needed for a thorough comparison.
Myasthenia Gravis vs. Organophosphate Poisoning - A Detailed Comparison
Both conditions affect the neuromuscular junction (NMJ) and involve acetylcholine (ACh) signaling, but they are mechanistically opposite and require very different management.
Quick Conceptual Summary
| Myasthenia Gravis | OP Poisoning | |
|---|---|---|
| Core defect | Too few AChRs (postsynaptic) | Too much ACh (AChE irreversibly inhibited) |
| ACh level at NMJ | Normal | Markedly elevated |
| ACh receptor | Blocked/destroyed by antibodies | Normal, but overstimulated |
| AChE | Normal | Irreversibly inhibited |
| Net effect | ACh cannot act effectively | ACh acts excessively and persistently |
1. Pathophysiology
Myasthenia Gravis
- Autoimmune - IgG antibodies target the alpha-1 subunit of nicotinic AChR on the postsynaptic muscle membrane.
- Antibodies cause: receptor blockade, accelerated receptor degradation, and complement-mediated destruction of postsynaptic folds.
- Result: 70-90% reduction in functional AChRs. End-plate potential (EPP) is barely above or below firing threshold. Safety factor of NMJ transmission is severely reduced.
- Presynaptic nerve and ACh release are completely normal.
- Clinically: fatigable weakness that worsens with use and improves with rest. - Bradley and Daroff's Neurology
Organophosphate Poisoning
- OPs (insecticides, nerve agents) irreversibly bind to and inactivate acetylcholinesterase (AChE) at all cholinergic synapses - NMJ, autonomic ganglia, and CNS.
- AChE cannot break down ACh, so ACh accumulates in the cleft and continues stimulating all cholinergic receptors persistently.
- The enzyme undergoes "aging" (irreversible covalent phosphorylation), making the inhibition permanent unless treated early with pralidoxime.
- Carbamates (e.g., neostigmine, pyridostigmine) cause similar but reversible inhibition - they do NOT undergo aging.
- Clinically: features of ACh excess at three receptor sites simultaneously. - Lippincott Illustrated Reviews: Pharmacology; Adams and Victor's Neurology
2. Clinical Features
The symptoms of OP poisoning are best organized by receptor type:
MUSCARINIC Effects (smooth muscle, glands) - Mnemonic: DUMBELS / SLUDGE
| Feature | OP Poisoning | MG |
|---|---|---|
| Miosis (pinpoint pupils) | ✅ Yes | ❌ No |
| Salivation / hypersecretion | ✅ Yes | ❌ No |
| Lacrimation | ✅ Yes | ❌ No |
| Bronchospasm / bronchorrhea | ✅ Yes | ❌ No (though may get aspiration) |
| Bradycardia | ✅ Yes | ❌ No |
| Diarrhea, urinary incontinence | ✅ Yes | ❌ No |
| Emesis, abdominal cramps | ✅ Yes | ❌ No |
| Sweating | ✅ Yes | ❌ No |
NICOTINIC Effects (NMJ and autonomic ganglia)
| Feature | OP Poisoning | MG |
|---|---|---|
| Muscle weakness | ✅ Yes (depolarizing blockade) | ✅ Yes (insufficient EPP) |
| Fasciculations | ✅ Yes (NMJ over-activation) | ❌ No |
| Tachycardia / hypertension | ✅ Yes (ganglionic) | ❌ No |
| Mydriasis (can occur) | ✅ (ganglionic > muscarinic) | ❌ No |
CNS Effects (OP only - crosses BBB)
| Feature | OP Poisoning | MG |
|---|---|---|
| Anxiety, restlessness | ✅ Yes | ❌ No |
| Seizures | ✅ Yes | ❌ No |
| Coma | ✅ Yes (severe cases) | ❌ No |
| LOC / confusion | ✅ Yes | ❌ No |
MG has no autonomic or CNS features - it is purely a peripheral skeletal muscle weakness problem. OP poisoning causes a pan-cholinergic syndrome affecting everything. - Lippincott Illustrated Reviews: Pharmacology
3. The Critical Overlap - and How to Distinguish Them
Both conditions cause muscle weakness - this is the main potential point of confusion. However:
| Distinguishing Feature | MG | OP Poisoning |
|---|---|---|
| Pupils | Normal (or slightly large from CN3 palsy) | Miosis (pinpoint) |
| Secretions | Dry | Wet (salivation, bronchorrhea, sweating) |
| Fasciculations | Absent | Present |
| Bradycardia | Absent | Present |
| CNS involvement | Absent | Present (seizures, coma) |
| Response to atropine | No benefit for weakness | Reverses muscarinic features |
| Anti-AChR antibodies | Present (80-90%) | Absent |
| Edrophonium (Tensilon) test | Improves strength | Worsens / no change (already too much ACh) |
| Course | Chronic, fluctuating | Acute, exposure history |
| Smell | Normal | May smell of garlic/petroleum |
- Tintinalli's Emergency Medicine; Goodman & Gilman's Pharmacological Basis of Therapeutics
4. Cholinergic Crisis vs. Myasthenic Crisis (Critical Distinction)
This is a clinically important comparison because overdose of pyridostigmine/neostigmine in MG can cause a cholinergic crisis that mimics MG worsening:
| Myasthenic Crisis | Cholinergic Crisis | |
|---|---|---|
| Cause | Disease exacerbation, infection, under-treatment | Excess AChE inhibitor (same drugs as MG treatment, or OP poisoning) |
| Weakness | Yes | Yes (depolarizing blockade of NMJ) |
| Secretions/bradycardia/miosis | Absent | Present |
| Fasciculations | Absent | Present |
| Edrophonium test | Improves strength | Worsens / causes fasciculations |
| Treatment | More AChE inhibitor, IVIG, plasma exchange | Withhold AChE inhibitor, give atropine |
The edrophonium (Tensilon) test is the bedside tool to distinguish these two: a small IV dose (2 mg, then up to 8 mg) reverses myasthenic crisis but worsens cholinergic crisis by further increasing ACh. - Goodman & Gilman's; Tintinalli's Emergency Medicine
5. Why Pyridostigmine Helps in MG but is Part of the Problem in OP Poisoning
| MG | OP Poisoning | |
|---|---|---|
| AChE status | Normal - just too few receptors | Already severely inhibited/destroyed |
| Adding AChE inhibitor | Beneficial - raises ACh to compensate for few receptors | Dangerous - piles excess ACh on top of already-maximal ACh, worsening depolarization blockade |
| Treatment goal | Increase ACh at the NMJ | Remove OP from AChE (pralidoxime), block muscarinic excess (atropine) |
In OP poisoning, giving pyridostigmine would be like pouring fuel on a fire - Goodman & Gilman's Pharmacological Basis of Therapeutics
6. Treatment Summary
| Treatment | MG | OP Poisoning |
|---|---|---|
| Pyridostigmine/Neostigmine | ✅ First-line symptomatic | ❌ Contraindicated |
| Atropine | Used only for AChI muscarinic side effects | ✅ Primary antidote (blocks muscarinic excess) |
| Pralidoxime (2-PAM) | Not used | ✅ Reactivates AChE if given before "aging" |
| Immunosuppressants | ✅ Steroids, azathioprine | ❌ Not relevant |
| Thymectomy | ✅ If thymoma or refractory disease | ❌ Not relevant |
| Plasma exchange / IVIG | ✅ For myasthenic crisis | ❌ Not relevant |
| Benzodiazepines | Not needed | ✅ For OP-related seizures |
Key Sources
- Bradley and Daroff's Neurology in Clinical Practice
- Goodman & Gilman's Pharmacological Basis of Therapeutics
- Lippincott Illustrated Reviews: Pharmacology
- Adams and Victor's Principles of Neurology, 12th Ed.
- Tintinalli's Emergency Medicine
How do pralidoxime and atropine work together in OP poisoning?
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Here is a full, textbook-grounded breakdown of how these two drugs work together.
Atropine + Pralidoxime in OP Poisoning: A Complementary Dual Strategy
The key concept is that neither drug alone is sufficient - they each address a completely different part of the toxidrome at a different level of the problem.
Step 1: Understand the Problem - Three Sites of Toxic ACh Excess
When an OP inhibits AChE, ACh accumulates and overstimulates receptors at three distinct locations:
| Site | Receptor Type | What Goes Wrong |
|---|---|---|
| Parasympathetic end organs (glands, heart, smooth muscle) | Muscarinic | DUMBELS syndrome |
| Neuromuscular junction (skeletal muscle) | Nicotinic (peripheral) | Fasciculations → paralysis |
| Brain | Nicotinic + Muscarinic (central) | Seizures, coma |
Atropine and pralidoxime divide this problem between them. Neither covers all three. - Tintinalli's Emergency Medicine
Atropine - "Block the Flood"
Mechanism: Atropine is a competitive antagonist at muscarinic receptors. It does not remove or reduce ACh - it simply outcompetes ACh for the receptor binding site, blocking its effects.
What it treats:
- Bronchospasm and bronchorrhea (most critical - the primary cause of death)
- Bradycardia and heart block
- Hypersalivation, lacrimation, sweating
- GI hypermotility (diarrhea, urination, vomiting)
- Miosis
What it does NOT treat:
- Skeletal muscle weakness, fasciculations, or respiratory muscle paralysis - these are nicotinic effects, and atropine has no activity at nicotinic receptors
- CNS seizures/coma - atropine does cross the BBB (it is a tertiary amine), so it does provide some central muscarinic blockade, but it cannot reverse the nicotinic and other CNS effects of ACh excess
Dosing approach:
- Start with 1-3 mg IV (0.05 mg/kg in children), doubling the dose every 5 minutes
- Titrate to the endpoint of drying of respiratory secretions and ease of breathing - NOT to heart rate or pupil size (those are unreliable endpoints)
- Severe poisoning may require 200-500 mg in the first hour, and up to 1 gram/day for weeks in extreme cases
- An infusion of 10-20% of the total cumulative atropinization dose per hour maintains effect
"Atropine is not active at nicotinic receptor sites and will not reverse skeletal muscle effects, such as respiratory muscle paralysis." - Rosen's Emergency Medicine
Pralidoxime (2-PAM) - "Restore the Enzyme"
Mechanism: Pralidoxime is an oxime - its key functional group is =NOH (the oxime group). This group has an extremely high affinity for the phosphorus atom of the OP-AChE complex.
Here is what happens at the molecular level:
- OPs phosphorylate the serine -OH group at the active site of AChE. This is the same serine that normally performs catalytic hydrolysis of ACh. With serine blocked, AChE is dead.
- Pralidoxime's oxime group (=NOH) competes with the serine -OH for the phosphorus atom.
- The oxime attacks the phosphorylated-enzyme complex, cleaves the phosphorus-serine bond, and regenerates the free serine -OH at the active site.
- AChE is now active again - it can once again hydrolyze ACh in the cleft.
The result: ACh is once again broken down normally, its concentration falls, and receptor overstimulation ceases.
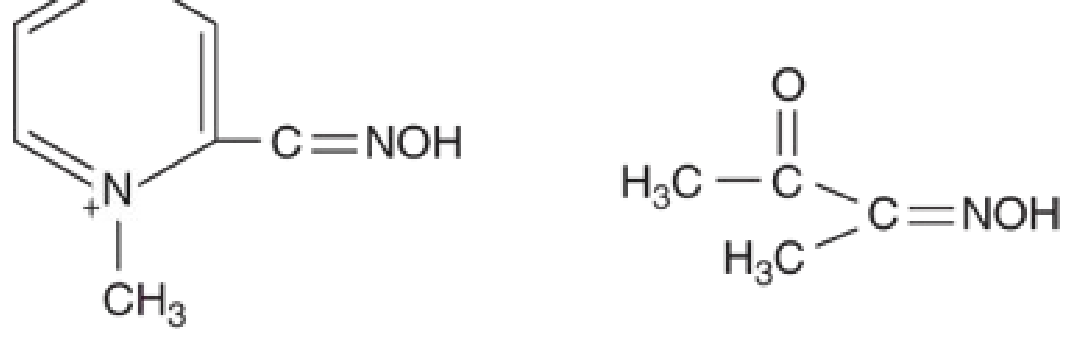
What pralidoxime treats (that atropine cannot):
- NMJ effects: fasciculations, muscle weakness, respiratory muscle paralysis
- Reverses the root cause of toxicity (ACh excess) rather than just blocking downstream receptors
What pralidoxime cannot treat:
- CNS effects - pralidoxime is a quaternary ammonium compound (note the N+ in the structure) and does NOT cross the blood-brain barrier. Central seizures and coma require benzodiazepines.
- It is most effective at the skeletal muscle NMJ, less so at other sites.
The Critical Limitation of Pralidoxime: "Aging"
This is the most important pharmacological concept for pralidoxime.
The aging process: After the OP binds to AChE, a secondary reaction progressively dealkylates the phosphoryl group, forming an even more stable bond with the serine residue. Once this "aging" is complete, the OP-AChE complex is irreversible and pralidoxime can no longer hydrolyze it - the window for enzyme rescue is closed.
Aging times by agent (clinically important):
| Agent | Approximate Aging Time |
|---|---|
| Sarin (GB) | ~5 hours |
| Tabun (GA) | ~14 hours |
| VX | ~40 hours |
| Soman (GD) | 2-6 minutes (extremely rapid - pralidoxime nearly useless) |
| Common insecticides (parathion, malathion) | Hours to days |
This is why pralidoxime must be given as early as possible - ideally within hours of exposure. With soman specifically, the aging is so rapid that pralidoxime is essentially ineffective, which is why pyridostigmine is used as prophylaxis before anticipated soman exposure (to transiently occupy AChE active sites and protect them from soman binding). - Katzung's Basic and Clinical Pharmacology; Tintinalli's Emergency Medicine
The Third Drug: Benzodiazepines
Neither atropine nor pralidoxime adequately controls OP-induced seizures and CNS toxicity:
- Atropine has limited central penetration and doesn't address nicotinic CNS excess
- Pralidoxime doesn't cross the BBB
Benzodiazepines (diazepam, lorazepam, midazolam) are the drug of choice for OP-induced seizures, acting through GABA-A receptors to suppress excessive CNS excitation. - Tintinalli's Emergency Medicine
How the Three Work Together: Summary Diagram
OP Poisoning
│
▼
AChE inhibited → ACh accumulates
│
├──► Muscarinic receptors ──────────► ATROPINE (competitive blockade)
│ (glands, heart, smooth muscle) Endpoint: dry secretions
│
├──► Nicotinic NMJ receptors ────────► PRALIDOXIME (enzyme reactivation)
│ (skeletal/respiratory muscle) Give early, before aging
│
└──► CNS receptors ─────────────────► BENZODIAZEPINES (seizure control)
(seizures, coma)
Why You Need Both Atropine AND Pralidoxime
| Scenario | Atropine alone | Pralidoxime alone |
|---|---|---|
| Bronchospasm/secretions | ✅ Controls | ❌ Slow (needs enzyme to act) |
| Respiratory muscle paralysis | ❌ Cannot treat | ✅ Reverses |
| Bradycardia | ✅ Controls | ❌ Not direct |
| Reduces ACh at source | ❌ No (just blocks receptors) | ✅ Yes (restores AChE) |
| Works after aging | ✅ Yes (doesn't depend on enzyme) | ❌ No |
| Time pressure | None | Must give ASAP |
Atropine buys time and controls the immediately life-threatening secretions and bronchospasm. Pralidoxime addresses the underlying biochemical problem and restores the NMJ function that atropine cannot touch. - Katzung's Basic and Clinical Pharmacology; Rosen's Emergency Medicine
Key sources:
- Katzung's Basic and Clinical Pharmacology, 16th Ed.
- Rosen's Emergency Medicine
- Tintinalli's Emergency Medicine
- Lippincott Illustrated Reviews: Pharmacology
Create a quick reference guide on atropine dosing and endpoints
Asking for Preferences
This is a shared conversation. Sign in to Orris to start your own chat.