Discuss pathophysiology of shock
Reading File
Finding Sources
Searching PubMed
shock pathophysiology mechanisms
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a thorough discussion of the pathophysiology of shock, drawn from Schwartz's Principles of Surgery (11th ed.), Harrison's Principles of Internal Medicine (22nd ed.), and Miller's Anesthesia (10th ed.).
Pathophysiology of Shock
"Shock is the manifestation of the rude unhinging of the machinery of life." - Samuel V. Gross, 1872
1. Definition and Core Concept
At its most fundamental level, shock is failure to meet the metabolic needs of the cell, regardless of etiology. The common denominator across all types of shock is inadequate oxygen delivery (DO2) relative to cellular oxygen demand, leading to cellular hypoxia and energy failure.
Mathematically:
DO2 = Cardiac Output (CO) × Arterial Oxygen Content (CaO2)
Where:
- CO = Heart Rate (HR) × Stroke Volume (SV)
- SV is determined by Preload × Contractility / SVR (afterload)
- CaO2 (mL/dL) = (Hb × 1.34 × SaO2) + (PaO2 × 0.003)
Any disease process that impairs HR, preload, contractility, SVR, SaO2, or hemoglobin has the potential to reduce DO2 and precipitate shock. - Harrison's Principles of Internal Medicine 22E, p. 3026-3038
2. Cellular and Metabolic Consequences
When oxygen supply becomes insufficient, cells can no longer sustain aerobic metabolism:
- Normally: glucose → pyruvate → enters mitochondria → oxidative phosphorylation → ATP (efficient)
- In shock: pyruvate is shunted to anaerobic glycolysis → lactate + only 2 ATP per glucose (vs. 36-38 via aerobic pathway)
The consequences of ATP depletion cascade rapidly:
| ATP-dependent process | Consequence of failure |
|---|---|
| Na+/K+ ATPase pump (uses 20-80% of cellular ATP) | Cell swells, loses ionic homeostasis |
| Intracellular pH regulation | Intracellular acidosis |
| Calcium extrusion | Ca2+ influx → activates phospholipases and proteases → cell death |
| Membrane integrity | Intracellular contents leak → activate systemic inflammatory cascades |
Cellular edema from Na+/K+ pump failure also restricts blood flow in adjacent capillaries - the "no-reflow" phenomenon - which can prevent reversal of ischemia even after adequate macroperfusion is restored. - Miller's Anesthesia 10e, p. 4812-4813
3. Phases of Shock
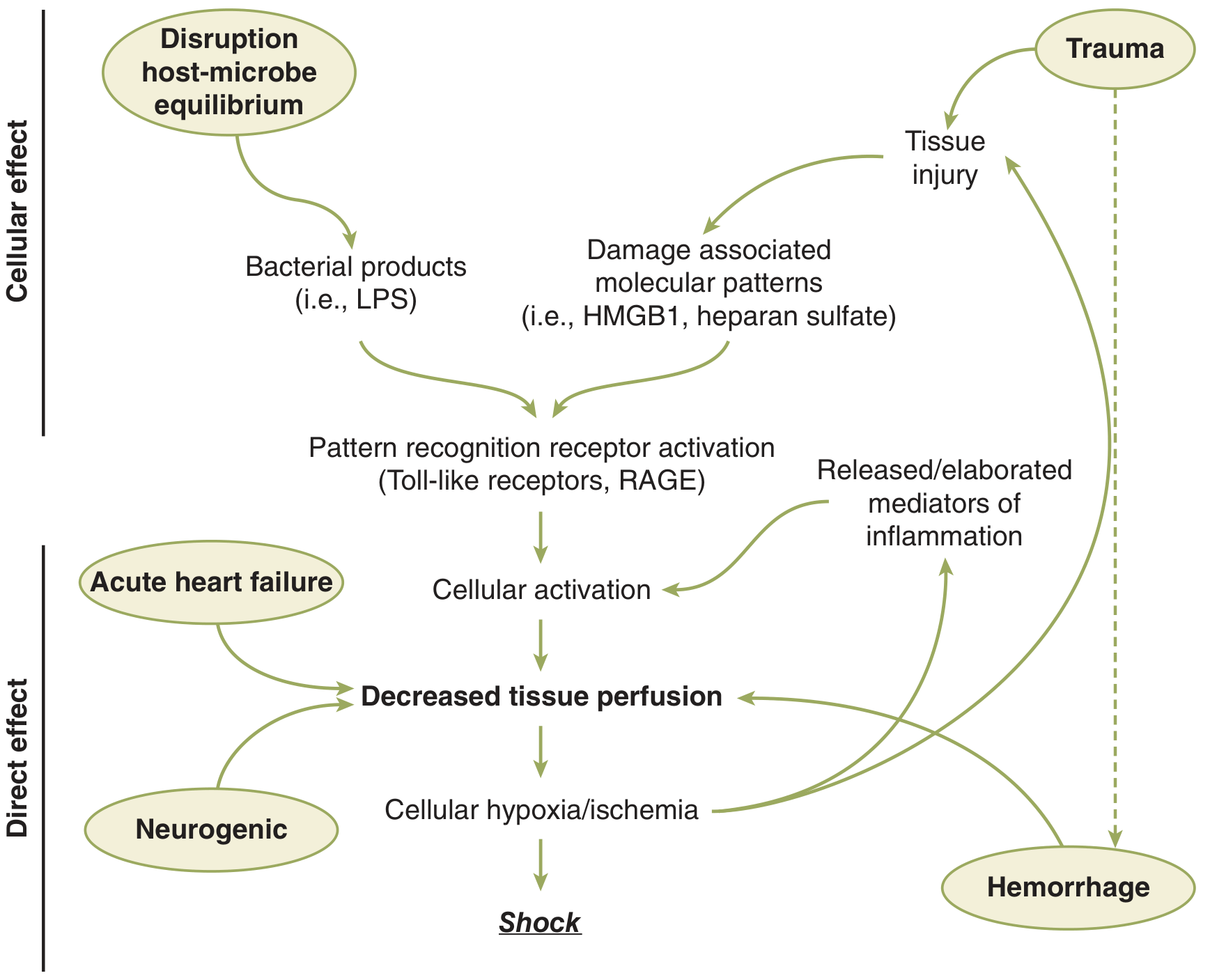
Shock progresses through three phases:
A. Compensated Phase
The body mounts a neuroendocrine response (detailed below) to maintain perfusion to the heart and brain. Hemodynamic parameters (BP, HR) may still be within normal or near-normal range. Cellular injury is ongoing but reversible.
B. Decompensation Phase
With continued hypoperfusion, the compensatory mechanisms are overwhelmed. Microcirculatory dysfunction, parenchymal tissue damage, and inflammatory cell activation perpetuate hypoperfusion. Ischemia/reperfusion injury exacerbates the initial insult.
C. Irreversible Phase
Extensive parenchymal and microvascular injury accumulates such that volume resuscitation fails to reverse the process. This leads to cardiovascular collapse and death. Clinically, this phase may develop insidiously and only be recognized in retrospect. - Schwartz's Principles of Surgery 11e, p. 2056-2069
4. The "Vicious Cycle" of Shock
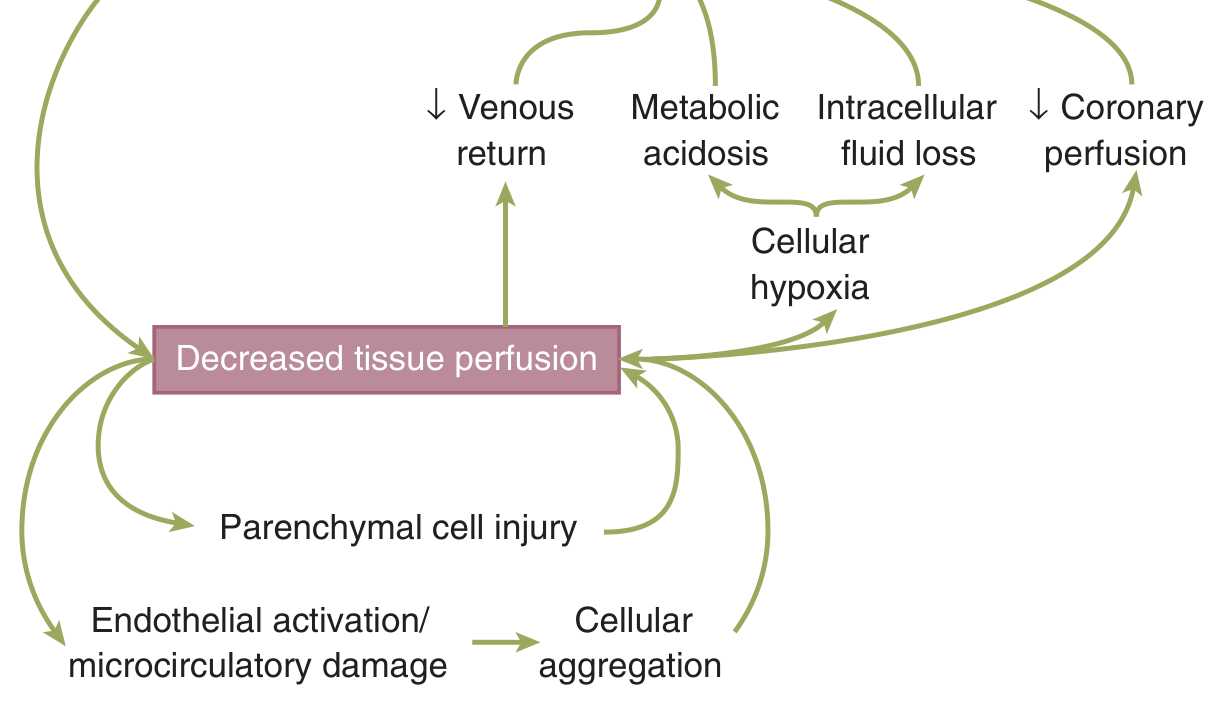
Decreased tissue perfusion triggers a self-reinforcing cycle:
- Cellular hypoxia → metabolic acidosis
- Metabolic acidosis depresses cardiac contractility → further reduced coronary perfusion
- Intracellular fluid loss (cellular swelling) further depletes intravascular volume
- Decreased venous return reduces preload → worsens cardiac output
- Endothelial activation and microcirculatory damage → cellular aggregation → capillary sludging → worsened perfusion
5. Neuroendocrine Response
The neuroendocrine stress response is the body's primary compensatory mechanism. Its goal is to maintain perfusion to the heart and brain at the expense of other organ systems.
Afferent Signals
Multiple sensors detect the shock state and relay signals to the CNS:
- Baroreceptors in the atria (low-pressure, detect volume), aortic arch and carotid bodies (high-pressure, detect pressure/stretch) - loss of inhibitory input dis-inhibits the ANS, activating sympathetic outflow
- Chemoreceptors in aorta and carotid bodies - detect hypoxia, hypercapnia, and acidemia
- Pain receptors - spinothalamic tracts activate the hypothalamic-pituitary-adrenal (HPA) axis
Efferent Responses
| Mediator | Source | Effect |
|---|---|---|
| Catecholamines (epinephrine, norepinephrine) | Adrenal medulla, sympathetic terminals | Vasoconstriction, ↑ HR, ↑ contractility |
| Renin → Angiotensin II → Aldosterone | Kidney / adrenal cortex | Na+ and water retention, vasoconstriction |
| ADH (vasopressin) | Posterior pituitary | Water retention, vasoconstriction |
| Cortisol | Adrenal cortex | Gluconeogenesis, potentiates catecholamines |
| Glucagon, Growth Hormone | Pancreas/pituitary | Metabolic substrate mobilization |
These responses together expand intravascular volume, increase vascular tone, and optimize cardiac output to sustain cerebral and coronary perfusion. - Schwartz's Principles of Surgery 11e, p. 2072-2084
6. Inflammatory and Immune Response
Beyond the neuroendocrine response, shock triggers a profound systemic inflammatory response:
Danger Signals
- Ischemic cells release Damage-Associated Molecular Patterns (DAMPs) - mitochondrial DNA, HMGB1, heparan sulfate, formyl peptides
- Bacterial translocation (especially in septic or prolonged shock) introduces PAMPs (e.g., LPS)
- These activate pattern recognition receptors - Toll-like receptors (TLRs) and RAGE - triggering cellular activation
Inflammatory Mediators Released
Ischemic cells produce a cascade of mediators: prostacyclin, thromboxane, prostaglandins, leukotrienes, endothelin, complement, interleukins, and tumor necrosis factor (TNF).
SIRS and Immune Paradox
The traditional view was:
- SIRS (pro-inflammatory) → CARS (Compensatory Anti-Inflammatory Response Syndrome) → recovery
More recent genomic research (the "Glue Grant") revealed a more nuanced picture:
- Both pro-inflammatory and anti-inflammatory innate immunity genes are upregulated simultaneously
- Adaptive immunity genes are concurrently downregulated
- Some patients exhibit amplified inflammatory responses that are slow to resolve - Miller's Anesthesia 10e, p. 4824-4828
7. Organ-Specific Responses
Brain
- Prime trigger of the neuroendocrine response
- Regional glucose uptake shifts during shock
- Reflexes and cortical electrical activity become depressed with hypotension - reversible with mild hypoperfusion, but permanent with prolonged ischemia
Heart
- Maintained perfusion as long as possible via coronary vasodilation
- Severe acidosis and hypoxia eventually depress myocardial contractility, closing the vicious cycle
Kidney
- Maintains GFR via selective vasoconstriction, shunting blood to medulla and deep cortex
- Prolonged hypotension → renal cell hibernation → tubular epithelial necrosis → acute kidney injury (AKI)
- Adrenal insufficiency occurs in up to 86% of severe hemorrhagic shock patients (observed in an n=59 cohort)
Liver
- Complex microcirculation susceptible to reperfusion injury
- Contributes to the inflammatory response and glucose dysregulation
- Failure of hepatic synthetic function is almost always lethal
Skeletal Muscle
- Metabolically less active; tolerates ischemia better than visceral organs
- Its large mass generates substantial lactic acid and free radicals from ischemic cells
- Sustained ischemia → intracellular Na+ and water accumulation → further depletes vascular volume - Miller's Anesthesia 10e, p. 4822-4856
8. Microcirculatory Failure
Even when macrocirculatory parameters are restored, microcirculatory failure can persist:
- Cellular edema compresses capillaries → no-reflow phenomenon
- Endothelial swelling, cellular aggregation, and microvascular thrombosis reduce effective tissue perfusion
- Leukocyte-endothelial adhesion (mediated by selectins and integrins) promotes capillary plugging and endothelial injury
- Glycocalyx degradation - the endothelial glycocalyx is shed by hypoperfusion, worsening microvascular permeability and contributing to coagulopathy
9. Coagulopathy in Shock
Shock - particularly traumatic/hemorrhagic - produces Acute Traumatic Coagulopathy (ATC):
- Tissue hypoperfusion → thrombomodulin-thrombin complex → generation of Activated Protein C (APC) → inactivates Factors Va and VIIIa → impaired fibrin formation
- Endothelial activation and injury supports a DIC-like consumptive picture
- Elevated plasmin relative to thrombin → hyperfibrinolytic state
- Degradation of the endothelial glycocalyx by hypoperfusion further amplifies coagulopathy
- Iatrogenic factors: hypothermia and large-volume crystalloid resuscitation worsen coagulopathy (patients receiving >4L crystalloid pre-hospital are at increased risk regardless of lactate level) - Miller's Anesthesia 10e, p. 4858-4870
10. Classification Summary (Hemodynamic Profiles)
| Type | Preload (CVP/PCWP) | CO/CI | SVR | Example |
|---|---|---|---|---|
| Hypovolemic | ↓ | ↓ | ↑ | Hemorrhage, dehydration |
| Cardiogenic | ↑ | ↓ | ↑ | MI, severe HF |
| Distributive | ↓/normal | ↑ | ↓↓ | Sepsis, anaphylaxis, neurogenic |
| Obstructive | ↑ | ↓ | ↑ | PE, cardiac tamponade, tension pneumothorax |
Each type has a distinct hemodynamic fingerprint corresponding to the specific variable in the DO2 equation that is primarily impaired. - Harrison's Principles of Internal Medicine 22E, p. 3047
Key Summary
The pathophysiology of shock is a progression from inadequate oxygen delivery → cellular energy failure → loss of ionic homeostasis → cell death → systemic inflammation → organ failure. The neuroendocrine response is the primary adaptive mechanism, but when overwhelmed, the resulting inflammatory cascade and microcirculatory failure create a self-amplifying vicious cycle. Without timely intervention, shock progresses from a compensated to an irreversible state, culminating in multi-organ failure and death.
Sources: Schwartz's Principles of Surgery 11e | Harrison's Principles of Internal Medicine 22e | Miller's Anesthesia 10e
Blood supply of heart and discuss cardiac causes of sudden natural death forensic notes
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material from the textbooks. Let me compose the full answer covering both parts: blood supply of the heart and cardiac causes of sudden natural death from a forensic perspective.
Blood Supply of the Heart & Cardiac Causes of Sudden Natural Death (Forensic Notes)
PART I: BLOOD SUPPLY OF THE HEART
Overview
The heart is supplied by two main coronary arteries - the Left Coronary Artery (LCA) and the Right Coronary Artery (RCA) - both arising from the aortic sinuses of Valsalva, just above the aortic valve. They are the first branches of the aorta and fill predominantly during diastole, when myocardial wall tension is lowest.
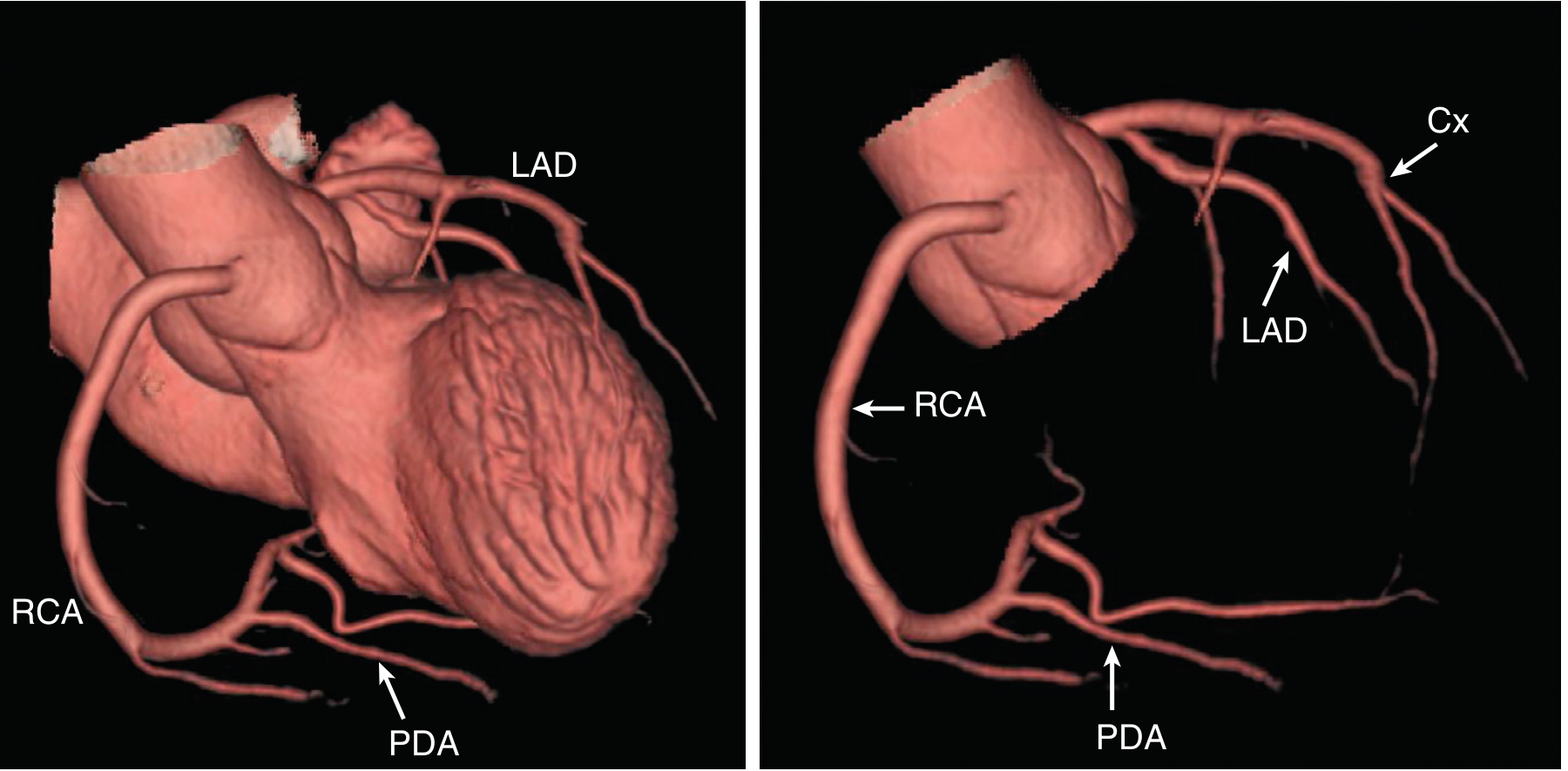
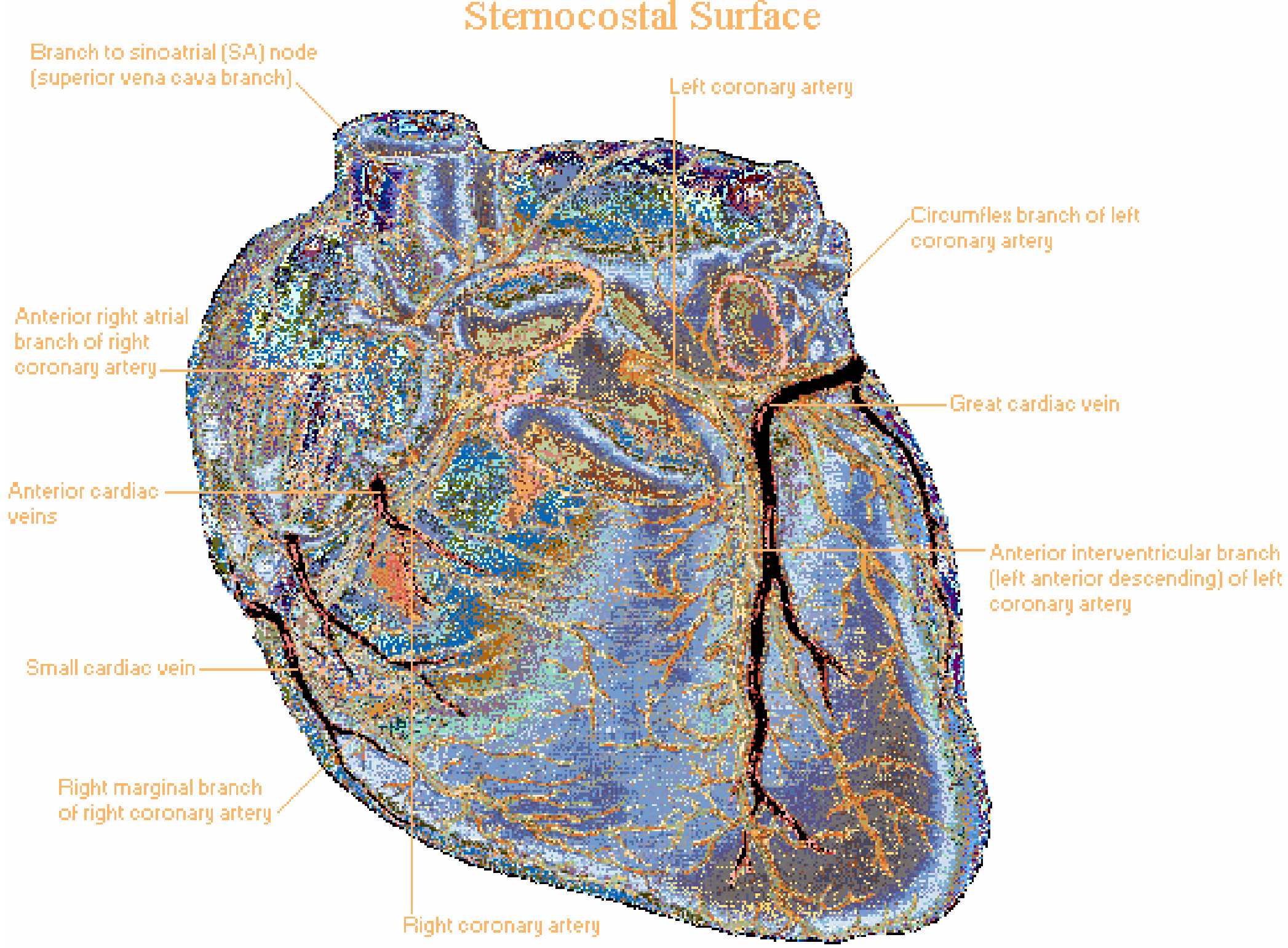
A. Left Coronary Artery (LCA)
Origin: The ostium of the LCA arises from the left sinus of Valsalva, near the sinotubular ridge. It runs as the left main coronary artery (LMCA) for a short distance before bifurcating (and occasionally trifurcating).
Main Divisions:
1. Left Anterior Descending Artery (LAD)
- Runs in the anterior interventricular groove toward (and sometimes around) the LV apex
- Branches:
- Septal perforators - supply the anterior 2/3 of the interventricular septum
- Diagonal branches - supply the anterior LV free wall
- Territory: Anterior wall of LV, anterior interventricular septum, LV apex (usually)
- Forensic importance: "Widow maker" - occlusion causes massive anterior MI
2. Circumflex Artery (Cx)
- Continues laterally in the atrioventricular (AV) groove after its right-angle origin from the left main
- Gives rise to obtuse marginal (OM) branches over the lateral to posterolateral LV free wall
- In left-dominant circulation (10-20% of people): the Cx gives rise to the posterior descending artery (PDA) and supplies the inferior LV
- Territory: Lateral wall and posterolateral LV wall; SA node in ~40% of people
Trifurcation (Ramus Intermedius)
In ~30% of individuals, the left main trifurcates into LAD, Cx, and an intermediate branch (ramus intermedius), which supplies the lateral LV wall.
B. Right Coronary Artery (RCA)
Origin: Arises from the right sinus of Valsalva. It travels in the right AV groove toward the inferior surface of the heart.
Branches along its course:
- Conus branch - first branch; supplies the right ventricular outflow tract (RVOT); collateral to LAD via Vieussens' ring
- SA node artery - supplies the sinoatrial node in ~60% of people (in the remaining 40%, SA nodal artery comes from the Cx)
- Right ventricular branches - supply the free wall of RV
- Acute marginal (right marginal) branch - large branch along the right heart border
- AV nodal artery - in right-dominant circulation, arises near the crux of the heart; supplies the AV node and bundle of His
- Posterior Descending Artery (PDA) - in right-dominant circulation (~80% of people), arises from the RCA; runs in the inferior interventricular groove and supplies the inferior 1/3 of the interventricular septum and inferior LV wall
Territory: Right ventricle free wall, inferior wall of LV, inferior septum (in right-dominant), SA node (60%), AV node (in right-dominant)
C. Coronary Dominance
| Dominance | PDA origin | Prevalence |
|---|---|---|
| Right dominant | RCA | ~80% |
| Left dominant | Circumflex | ~10-15% |
| Co-dominant/Balanced | Both | ~5-10% |
The dominant artery supplies the posterior descending artery and also gives off the AV nodal artery. Occlusion of the dominant vessel therefore threatens both the inferior myocardium and the conduction system.
D. Venous Drainage
The venous drainage follows the arterial supply in a general sense:
| Vein | Drains | Empties into |
|---|---|---|
| Great cardiac vein | Accompanies LAD | Coronary sinus |
| Middle cardiac vein | Accompanies PDA | Coronary sinus |
| Small cardiac vein | Right heart border | Coronary sinus |
| Anterior cardiac veins | Anterior RV wall | Directly into right atrium |
| Thebesian veins (venae cordis minimae) | Myocardium | Directly into cardiac chambers |
The coronary sinus drains ~75% of coronary venous blood and empties into the right atrium.
E. Nerve Supply
- The heart receives autonomic supply via the cardiac plexus (superficial and deep), formed by sympathetic fibers (from T1-T5 cervicothoracic ganglia) and parasympathetic fibers (vagus nerve, CN X)
- Sympathetic: increases HR, contractility, conduction velocity; dilates coronary arteries
- Parasympathetic: slows HR, reduces conduction; minimal direct effect on ventricular myocardium
PART II: CARDIAC CAUSES OF SUDDEN NATURAL DEATH (FORENSIC PERSPECTIVE)
Definition
Sudden natural death in forensic pathology is defined as an unexpected death from natural causes, typically occurring within 1 hour of symptom onset (or within 24 hours of the deceased last being seen alive and well). "Cardiac" SND refers to cases attributable to a cardiac lesion at autopsy.
Cardiac causes account for the majority (~70-80%) of all sudden natural deaths, making this the single most important category in forensic pathology.
Forensic Classification of Cardiac SND
1. Ischemic Heart Disease (IHD) / Coronary Artery Disease - MOST COMMON (65-80%)
CAD is the leading cause of sudden cardiac death. Key forensic points:
a) Atherosclerosis with Critical Stenosis
- The most common finding at autopsy is severe chronic atherosclerotic disease with fixed critical stenoses (≥75% luminal narrowing) without acute plaque disruption
- SCD can be the first clinical manifestation of IHD - the patient may have had no prior symptoms
- Mechanism: Severe, chronic ischemia creates electrical irritability of the myocardium distant from the conduction system → lethal arrhythmia (ventricular fibrillation or asystole)
b) Acute Plaque Rupture/Thrombosis
- Found in only 10-20% of SCD cases at autopsy
- Coronary angiography (or coronary dissection at autopsy) shows thrombotic occlusion in approximately 50% of cases overall
- 80-90% of SCD victims show no enzymatic or ECG evidence of myocardial necrosis, even when IHD is the cause - this is critical forensically (no classic MI pattern necessary)
c) Remote MI
- Healed remote MIs are present in ~40% of SCD cases - old infarct scar predisposes to re-entry arrhythmias
- Subendocardial myocyte vacuolization (contraction band necrosis pattern) is a marker of severe chronic ischemia, often found without full infarction
Forensic Autopsy Approach:
- Cross-section coronary arteries at 3-5 mm intervals throughout their entire length
- Document degree of stenosis, plaque morphology, presence of thrombus, calcification
- Document weight of heart (normal adult ~300-350g), wall thickness (LV >15 mm = hypertrophy)
- Microscopic sections of myocardium essential: necrosis, fibrosis, vacuolization
2. Hypertensive Heart Disease
- Increased cardiac mass is an independent risk factor for SCD
- In some young individuals and athletes, hypertensive LV hypertrophy or unexplained increased cardiac mass may be the only pathologic finding at autopsy
- Mechanism: Hypertrophied myocardium is prone to ischemia (increased O2 demand, reduced subendocardial perfusion reserve) and fibrosis → arrhythmic substrate
- In high-resource countries where IHD prevalence is declining, SCD increasingly occurs in hearts that are hypertrophic and fibrotic from hypertension, obesity, or cocaine use - Robbins & Kumar, p. 1511
3. Cardiomyopathies
a) Hypertrophic Cardiomyopathy (HCM)
- A leading cause of SCD in young people and athletes (aged <35 years)
- Autosomal dominant mutations in sarcomeric proteins (most commonly beta-myosin heavy chain, myosin-binding protein C)
- Pathology: Asymmetric septal hypertrophy, myocyte disarray, interstitial fibrosis
- Mechanism: Outflow tract obstruction, diastolic dysfunction, and arrhythmic substrate from disarray and fibrosis
- Forensic finding: Disproportionate septal hypertrophy (septal:free wall ratio >1.3), disorganized myofiber arrangement on microscopy
b) Dilated Cardiomyopathy (DCM)
- Dilated, poorly contracting ventricles; causes include viral myocarditis, idiopathic, alcohol, genetic
- SCD via ventricular arrhythmias from stretched, fibrotic myocardium
c) Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
- Fibro-fatty replacement of the RV myocardium
- Classic cause of SCD in young athletes, especially during exercise
- Forensic finding: RV dilation with fatty infiltration, thinned RV wall - requires histological confirmation
4. Channelopathies (Structurally Normal Hearts)
These account for 5-10% of SCD, especially in young individuals. The heart may appear grossly normal at autopsy - a critical forensic challenge.
| Condition | Mechanism | ECG pattern in life |
|---|---|---|
| Long QT Syndrome (LQTS) | Mutation in K+ or Na+ channels → delayed repolarization → Torsades de Pointes | Prolonged QTc |
| Brugada Syndrome | SCN5A Na+ channel mutation → VF, predominantly nocturnal | Coved ST elevation in V1-V3 |
| Short QT Syndrome | Gain-of-function K+ channel → shortened repolarization | Short QTc |
| Catecholaminergic Polymorphic VT (CPVT) | RyR2 mutation → Ca2+ channel dysregulation | Normal at rest; VT on exercise |
Forensic challenge: These cases require molecular autopsy (postmortem genetic testing) as no structural abnormality is identifiable. Cause of death may be certified as "unascertained" or "sudden arrhythmic death syndrome (SADS)" without genetic testing.
5. Myocarditis
- Inflammatory infiltrate of the myocardium, most commonly viral (Coxsackievirus B, adenovirus, SARS-CoV-2, etc.)
- Can cause fatal arrhythmias even before clinical diagnosis
- Forensic significance: Myocarditis can occur in young, apparently healthy individuals
- Autopsy finding: Edematous, flabby heart; microscopy shows lymphocytic infiltrate with myocyte necrosis (Dallas criteria)
- Sarcoid myocarditis is a less common but important cause of SCD via VF
6. Valvular Heart Disease
- Aortic stenosis: Critical AS (valve area <1 cm²) can cause SCD via decreased cardiac output or arrhythmia, particularly on exertion
- Mitral valve prolapse (MVP): Most cases are benign; however, myxomatous MVP with thickened, redundant leaflets and papillary muscle fibrosis can cause SCD, particularly in young women
- Infective endocarditis: Vegetations can embolize to coronary arteries or cause sudden valvular destruction
7. Congenital Coronary Artery Anomalies
- Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA): causes myocardial ischemia from birth
- Anomalous origin from wrong sinus: especially the left coronary from the right sinus (with an interarterial course between the aorta and pulmonary trunk) - may be compressed during exercise → SCD in young athletes
- These anomalies occur in 1-2% of the population; some are only discovered at autopsy
- Forensic approach: Careful examination of coronary ostia and their relationship to the aortic valve is mandatory at every cardiac autopsy
8. Aortic Dissection / Rupture
- Type A dissection can extend to the coronary ostia → acute MI and tamponade → SCD
- Marfan syndrome and bicuspid aortic valve are predisposing conditions
- Forensic finding: Hemopericardium (cardiac tamponade), intimal tear in ascending aorta
9. Cardiac Tamponade
- Sudden accumulation of blood in pericardial sac (200-300 mL can be fatal)
- Causes: Aortic dissection, ventricular rupture post-MI, trauma (forensic must exclude), tumor
- Forensic note: Must distinguish natural (hemopericardium from MI rupture, dissection) vs. traumatic tamponade
Forensic Autopsy Protocol for Cardiac SND
- Gross examination: Heart weight, ventricular wall thickness (LV, RV, septum), chamber size, epicardial fat, valves, chordae tendineae
- Coronary arteries: Serial cross-sections at 3-5 mm intervals; document stenosis %, thrombus, dissection, calcification
- Myocardium: Slice in short-axis planes (bread-loaf technique); document infarct age (acute, healing, healed), fibrosis, hypertrophy
- Histology (mandatory): Sections from LV, RV, septum, SA node, AV node; stain with H&E, Masson's trichrome (fibrosis), Congo red (amyloid) as needed
- Toxicology: Cocaine, amphetamines, and other cardiotoxic substances must be excluded
- Molecular autopsy: Genetic panel (KCNQ1, KCNH2, SCN5A, RYR2, MYBPC3, MYH7, etc.) in structurally normal hearts, especially in the young
Summary Table: Cardiac Causes of Sudden Natural Death
| Cause | Age group | Key finding at autopsy | Mechanism |
|---|---|---|---|
| Coronary atherosclerosis (IHD) | Middle-aged/elderly | Critical stenosis, ± thrombus, ± healed MI | VF from ischemic irritability |
| Hypertensive heart disease | Adults | LVH, no other pathology | Arrhythmia from hypertrophied, fibrotic heart |
| Hypertrophic cardiomyopathy | Young, athletes | Asymmetric septal hypertrophy, myocyte disarray | VT/VF |
| ARVC | Young athletes | Fibro-fatty RV replacement | VT/VF on exertion |
| Dilated cardiomyopathy | Any | Dilated, flabby heart | VT/VF |
| Myocarditis | Young | Lymphocytic infiltrate + myonecrosis | VT/VF |
| Channelopathies | Young | Structurally normal heart | Primary electrical instability |
| Mitral valve prolapse | Young women | Myxomatous thickened leaflets | VT/VF |
| Congenital coronary anomaly | Young, athletes | Anomalous coronary origin | Ischemia → VF |
| Aortic stenosis | Elderly | Calcific AS, LVH | Low-output or arrhythmia |
| Cardiac tamponade | Any | Hemopericardium | Obstructive shock |
Key Forensic Principle: The mechanism of death in cardiac SND is almost universally a lethal arrhythmia (ventricular fibrillation, ventricular tachycardia, or asystole). The underlying cause (structural lesion) must be established at autopsy. When no structural cause is found - particularly in the young - molecular autopsy for channelopathies is strongly recommended.
Sources: Robbins & Kumar Basic Pathology, p. 723-749 | Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1505-1525 | Textbook of Clinical Echocardiography, p. 2434-2458 | Fuster and Hurst's The Heart 15e, p. 3120-3128 | Netter's Atlas of Human Anatomy
Notes
Asking for Preferences
Given the context of the previous conversation, here are clean, exam-ready revision notes covering both topics discussed:
BLOOD SUPPLY OF THE HEART - Revision Notes
Coronary Arteries - Origin
- Both arise from aortic sinuses of Valsalva, just above the aortic valve
- Fill predominantly during diastole (when myocardial wall tension is lowest)
Left Coronary Artery (LCA)
Origin: Left sinus of Valsalva → short Left Main → bifurcates into:
LAD (Left Anterior Descending)
- Runs in anterior interventricular groove
- Branches: Septal perforators (anterior 2/3 septum) + Diagonal branches (anterior LV wall)
- Territory: Anterior LV, anterior septum, apex
- "Widow maker" - occlusion = massive anterior MI
Circumflex (Cx)
- Runs in AV groove laterally
- Gives Obtuse Marginal (OM) branches to lateral/posterolateral LV
- In left-dominant (10-20%): gives rise to PDA
- Supplies SA node in ~40% of people
Ramus Intermedius
- Present in ~30%; trifurcation of left main → supplies lateral LV
Right Coronary Artery (RCA)
Origin: Right sinus of Valsalva → travels in right AV groove inferiorly
| Branch | Territory |
|---|---|
| Conus branch (1st branch) | RV outflow tract; collateral to LAD via Vieussens' ring |
| SA nodal artery | SA node in ~60% of people |
| Right ventricular branches | RV free wall |
| Acute (right) marginal branch | Right heart border |
| AV nodal artery | AV node + bundle of His (in right dominant) |
| PDA (posterior descending) | Inferior septum, inferior LV wall |
Coronary Dominance
| Type | PDA from | % Population |
|---|---|---|
| Right dominant | RCA | ~80% |
| Left dominant | Circumflex | ~10-15% |
| Co-dominant | Both | ~5-10% |
Dominant artery = supplies both PDA and AV nodal artery
Venous Drainage
| Vein | Drains | Into |
|---|---|---|
| Great cardiac vein | Accompanies LAD | Coronary sinus |
| Middle cardiac vein | Accompanies PDA | Coronary sinus |
| Small cardiac vein | Right heart border | Coronary sinus |
| Anterior cardiac veins | Anterior RV | Directly into RA |
| Thebesian veins | Deep myocardium | Directly into chambers |
- Coronary sinus → drains ~75% venous blood → empties into RA
Arterial Supply - Regional Summary
| Region | Supplied by |
|---|---|
| Anterior LV wall | LAD (diagonal branches) |
| Anterior 2/3 septum | LAD (septal perforators) |
| Lateral LV | Circumflex (OM branches) |
| Inferior LV wall | PDA (from RCA in right dominant) |
| Inferior 1/3 septum | PDA |
| RV free wall | RCA |
| SA node | RCA (60%), Cx (40%) |
| AV node | RCA (80%), Cx (20%) |
CARDIAC CAUSES OF SUDDEN NATURAL DEATH - Forensic Notes
Definition
- Death from natural cardiac cause within 1 hour of symptom onset (or within 24 hours of last being seen alive/well)
- Cardiac causes = 70-80% of all sudden natural deaths
- Mechanism is almost always a lethal arrhythmia (VF, VT, asystole) - the structural lesion is the cause, arrhythmia is the mechanism
1. Ischemic Heart Disease (CAD) - MOST COMMON (65-80%)
Key Forensic Points:
- Most common finding = severe fixed atherosclerotic stenosis WITHOUT acute plaque rupture
- Acute plaque disruption/thrombosis found in only 10-20% of cases
- Coronary thrombosis found in ~50% on angiography/autopsy
- 80-90% show NO enzymatic or ECG evidence of myocardial necrosis - no MI pattern needed for diagnosis
- ~40% have healed remote MI (old infarct scar = re-entry arrhythmia substrate)
- Subendocardial myocyte vacuolization = chronic ischemia marker
Autopsy:
- Serial cross-sections of coronaries at 3-5 mm intervals (entire length)
- Document: % stenosis, thrombus, plaque calcification, acute vs chronic changes
- Heart weight, LV wall thickness (>15 mm = hypertrophy)
2. Hypertensive Heart Disease
- Increased cardiac mass = independent risk factor for SCD
- In some young individuals/athletes, LVH may be the only pathologic finding at autopsy
- Mechanism: ↑ O2 demand + reduced subendocardial perfusion + fibrosis → arrhythmic substrate
- Increasingly seen as IHD prevalence declines (obesity, cocaine, hypertension)
3. Cardiomyopathies
| Type | Key Features | Forensic Finding |
|---|---|---|
| HCM (Hypertrophic) | #1 cause SCD in young athletes | Asymmetric septal hypertrophy, myocyte disarray, fibrosis |
| DCM (Dilated) | Viral/idiopathic/alcohol/genetic | Dilated, flabby, heavy heart |
| ARVC | Fibro-fatty RV replacement; exercise-triggered | Fatty infiltration of RV wall (needs histology) |
- HCM: Septal:free wall ratio >1.3:1; autosomal dominant sarcomeric mutations
- ARVC: Particularly in young athletes; standard gross examination may miss - histology mandatory
4. Channelopathies - "Structurally Normal Heart" SCD
| Condition | Gene | Mechanism | Key Clue |
|---|---|---|---|
| Long QT (LQTS) | KCNQ1, KCNH2, SCN5A | Delayed repolarization → Torsades | Prolonged QTc in life |
| Brugada Syndrome | SCN5A | Na+ channel dysfunction → VF | Coved ST ↑ V1-V3; nocturnal |
| CPVT | RYR2, CASQ2 | Ca2+ leak → VT on exercise | Exercise-triggered, normal at rest |
| Short QT | KCNH2, KCNQ1 | Shortened repolarization | Short QTc |
Forensic challenge: Heart is grossly and histologically normal - cause of death = "SADS" (Sudden Arrhythmic Death Syndrome) without molecular autopsy Molecular autopsy (genetic panel) is mandatory in young SCD with no structural findings
5. Myocarditis
- Most commonly viral (Coxsackievirus B, adenovirus, SARS-CoV-2)
- Can be first presentation in apparently healthy young individuals
- Sarcoidosis - granulomatous myocarditis is an important cause of SCD in young/middle-aged
- Autopsy: Flabby, edematous heart; microscopy = lymphocytic infiltrate + myocyte necrosis (Dallas criteria)
6. Valvular Causes
| Valve lesion | Mechanism of SCD |
|---|---|
| Aortic stenosis (critical) | Low output on exertion, arrhythmia; calcified valve, LVH |
| Mitral valve prolapse | Myxomatous thickening + papillary fibrosis → VF (esp. young women) |
| Infective endocarditis | Coronary embolism, acute valvular destruction |
7. Congenital Coronary Anomalies
- Anomalous LCA from right sinus (interarterial course) = compression between aorta and PA during exercise → SCD in young athletes
- ALCAPA (Anomalous LCA from pulmonary artery) = ischemia from birth
- Occur in 1-2% of population; discovered at autopsy
- Coronary ostia examination is mandatory at every cardiac autopsy
8. Other Important Causes
| Cause | Forensic Notes |
|---|---|
| Aortic dissection Type A | Hemopericardium (tamponade), intimal tear in ascending aorta; Marfan's, bicuspid AV |
| Cardiac tamponade | 200-300 mL blood = fatal; distinguish natural (MI rupture, dissection) vs traumatic |
| Pulmonary embolism | Saddle embolism → acute RV failure → SCD; look for DVT source |
Forensic Autopsy Protocol for Cardiac SND
| Step | Action |
|---|---|
| 1. External | Body weight, habitus, stigmata of genetic syndromes (Marfan's) |
| 2. Heart weight | Normal adult ~300-350g; >500g = significant hypertrophy |
| 3. Gross heart | Chamber size, wall thickness (LV, RV, septum), epicardial appearance |
| 4. Coronary arteries | Serial 3-5 mm cross-sections entire length; % stenosis, thrombus |
| 5. Myocardium | Bread-loaf technique; age of infarct (acute/healing/healed), fibrosis |
| 6. Valves | Size, calcification, vegetations, myxomatous change |
| 7. Histology | H&E (all chambers + SA/AV node); Masson's trichrome (fibrosis); Congo red (amyloid) |
| 8. Toxicology | Cocaine, amphetamines, alcohol, cardiotoxic drugs |
| 9. Molecular autopsy | Genetic panel if structurally normal heart + young age |
Age-Based Pattern Recognition (Exam Tip)
| Age Group | Most Common Cause |
|---|---|
| <35 years (athletes) | HCM, ARVC, congenital coronary anomaly, myocarditis, channelopathy |
| 35-50 years | Coronary atherosclerosis, HCM, channelopathy |
| >50 years | Coronary atherosclerosis, hypertensive heart disease, aortic stenosis |
One-Liner Key Facts
- Mechanism of death in cardiac SND = always ventricular fibrillation/asystole
- Most common cause overall = CAD/atherosclerosis
- Most common cause in young athletes = HCM
- Structurally normal heart SCD = channelopathy (LQTS, Brugada, CPVT) → needs molecular autopsy
- SCD can be the first manifestation of IHD in up to 25% of cases
- No MI at autopsy ≠ no ischemic cause - 80-90% of SCD from IHD show no MI
- Old infarct scar found in ~40% of SCD cases
- Coronary thrombus found in only ~50% of fatal CAD cases
This is a shared conversation. Sign in to Orris to start your own chat.