Tell me about acute pyelonephritis for step 1
"pyelonephritis"[MeSH Terms] AND "diagnosis"[MeSH Terms]
Acute Pyelonephritis - Step 1 Review
Definition
- Robbins & Kumar Basic Pathology, p. 514
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 859
Pathogenesis - Routes of Infection
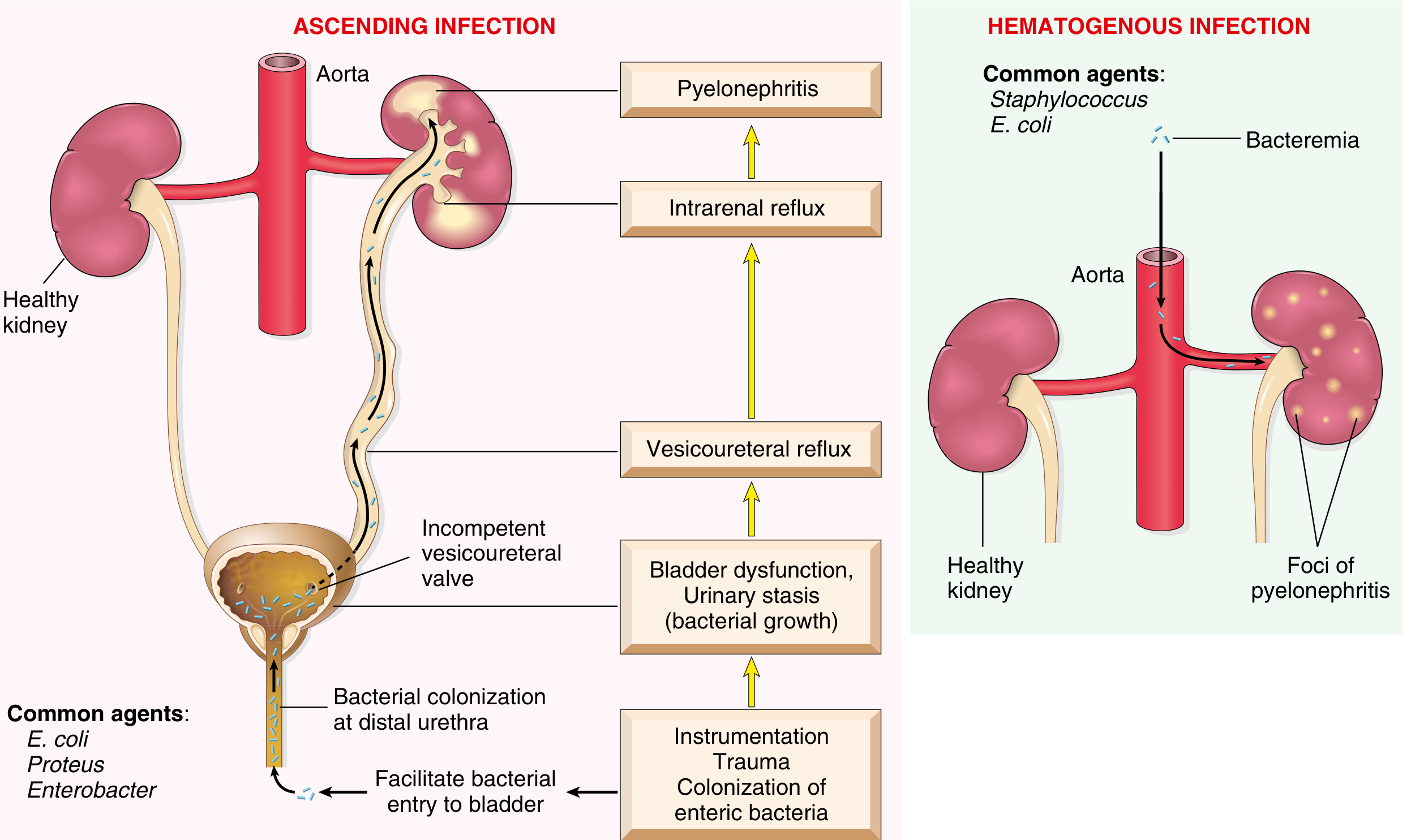
- Colonization of distal urethra/introitus by enteric bacteria (especially E. coli)
- Instrumentation, trauma, or sexual activity facilitates bladder entry
- Urinary stasis (obstruction, bladder dysfunction, neurogenic bladder, VUR) allows bacterial multiplication
- Vesicoureteral reflux (VUR) - incompetent ureterovesical valve - propels infected urine up to the renal pelvis and parenchyma via intrarenal reflux
- VUR present in 20-40% of children with UTI (usually congenital)
- Bacteremia (e.g., endocarditis, sepsis) seeds the kidney
- Common agents here: Staphylococcus aureus, E. coli
- Produces multifocal cortical abscesses rather than medullary/pelvic disease
Microbiology
| Organism | Notes |
|---|---|
| E. coli | Most common (80% of cases); possesses P pili that bind to P blood group antigen receptors on urothelium (virulence factor) |
| Klebsiella | Common, especially in diabetics |
| Proteus | Urease-producing - raises urinary pH, causes struvite stones |
| Enterobacter | Less common gram-negative rod |
| Pseudomonas | Associated with nosocomial/catheter infections |
| Staphylococcus | Hematogenous route; S. saprophyticus in young sexually active women |
- All gram-negative enteric bacilli are normal intestinal flora.
- Campbell Walsh Wein Urology, p. 3308
Risk Factors / Predisposing Conditions
- Female sex - short urethra close to rectum; most common in sexually active women
- Urinary tract obstruction - BPH, calculi, uterine prolapse, posterior urethral valves
- Vesicoureteral reflux (VUR)
- Pregnancy - uterine compression causes urinary stasis; 4-6% develop bacteriuria; 20-40% of untreated bacteriuria progresses to pyelonephritis
- Catheterization / urinary tract instrumentation
- Diabetes mellitus - immunosuppression + neurogenic bladder dysfunction
- Immunosuppression / immunodeficiency
- Preexisting renal scarring
Clinical Presentation
Fever + Flank/CVA (costovertebral angle) pain/tenderness + Dysuria
-
Systemic: abrupt onset of chills, fever (≥100.4°F / 38°C), malaise, nausea/vomiting
-
Urinary: dysuria, frequency, urgency (lower tract symptoms may also be present or absent)
-
Physical exam: costovertebral angle (CVA) tenderness to deep palpation
-
Can simulate GI illness with abdominal pain, nausea, vomiting
-
Usually unilateral - does NOT typically cause renal failure in uncomplicated cases
-
Robbins & Kumar Basic Pathology, p. 516
-
Campbell Walsh Wein Urology, p. 3294
Lab Findings
| Test | Finding | Significance |
|---|---|---|
| Urinalysis | Pyuria (many WBCs), bacteriuria | Core finding |
| WBC casts | Neutrophil-rich (pus) casts | Pathognomonic for kidney involvement (casts form only in tubules) |
| Urine culture | Positive (usually >10^5 CFU/mL) | Establishes diagnosis; ~20% have <10^5 CFU/mL |
| CBC | Leukocytosis with left shift (neutrophilia) | Systemic infection |
| Blood cultures | May be positive | Bacteremia/urosepsis |
| CRP/ESR | Elevated | Inflammatory markers |
| Creatinine | May be elevated in severe cases | AKI (uncommon in uncomplicated cases) |
Morphology / Pathology
- Discrete, raised, yellowish-white abscesses on the cortical surface
- May form large wedge-shaped areas of necrosis
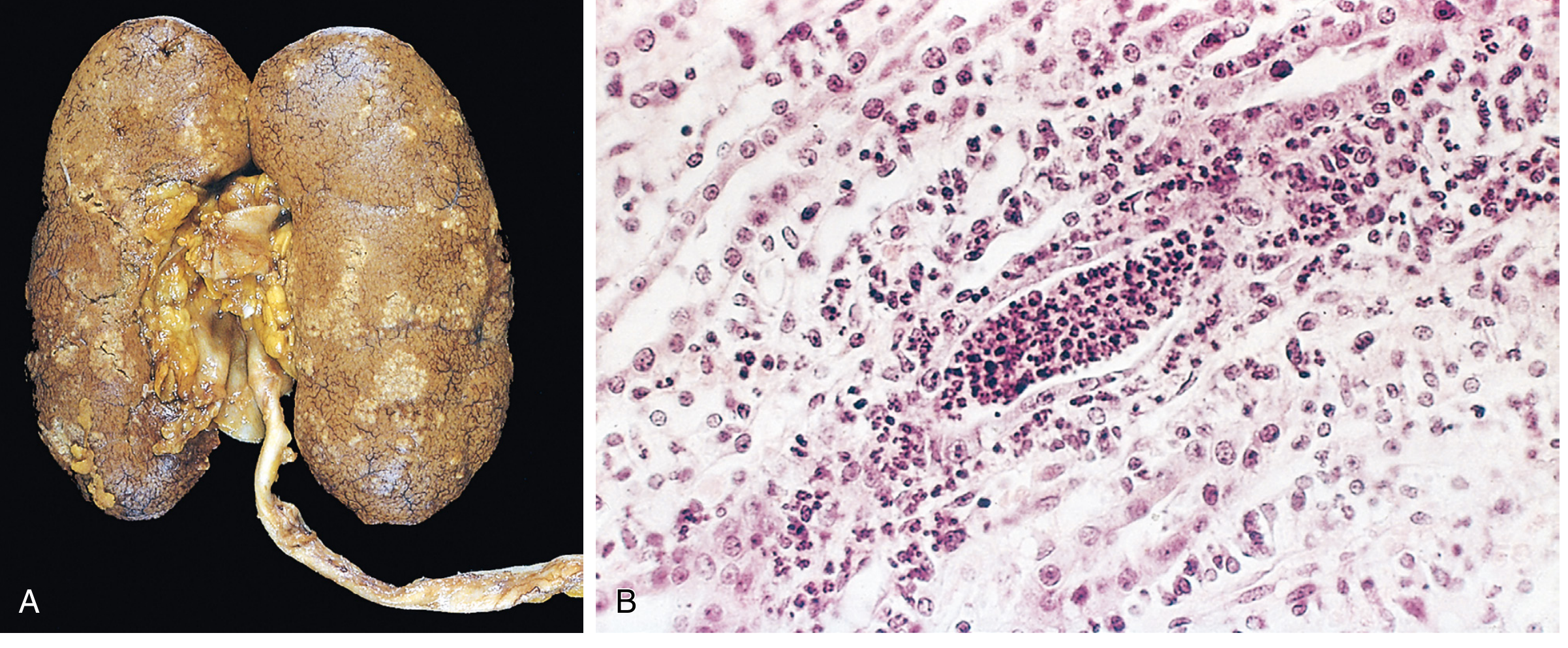
- Patchy interstitial suppurative inflammation
- Intratubular aggregates of neutrophils (neutrophilic tubulitis)
- Tubular injury with extension into the interstitium
- Glomeruli are relatively SPARED (characteristically resistant)
- Fungal infections (e.g., Candida) cause granulomatous interstitial inflammation instead
Complications
1. Papillary Necrosis
- Ischemic + suppurative necrosis of the tips/distal two-thirds of the renal papillae
- Classic triad: Diabetes + Urinary obstruction + Sickle cell disease (also analgesic nephropathy)
- Usually bilateral but can be unilateral
- Microscopy: coagulative necrosis of papillary tips with preserved tubular outlines
- May lead to acute renal failure if sloughed papillae cause obstruction
2. Pyonephrosis
- Total or near-complete urinary obstruction, especially high in the urinary tract
- Suppurative exudate fills the renal pelvis, calyces, and ureter with pus
- Unable to drain - medical emergency
3. Perinephric Abscess
- Extension of suppurative inflammation through the renal capsule into perinephric fat
- May not respond to antibiotics alone; requires drainage
Treatment (Step 1 Overview)
| Setting | Preferred Agents |
|---|---|
| Mild-moderate, outpatient | Fluoroquinolones (ciprofloxacin 5-7 days; levofloxacin 5-7 days) or TMP-SMX x 14 days |
| Hospitalized / severe | IV aminoglycoside (gentamicin) initial empiric therapy; transition to oral after 24-48 hrs of improvement |
| Pregnancy | IV cephalosporins or penicillins until afebrile, then 14 days total oral |
| Complicated/resistant | Meropenem, piperacillin-tazobactam, plazomicin |
- Nitrofurantoin and fosfomycin are NOT appropriate for pyelonephritis (insufficient renal tissue levels)
- Duration: 7-14 days total (5-7 days adequate for fluoroquinolones)
- Goldman-Cecil Medicine, p. 2979
Key Distinctions for Step 1
| Feature | Cystitis (Lower UTI) | Pyelonephritis (Upper UTI) |
|---|---|---|
| Fever/chills | Usually absent | Present |
| CVA tenderness | Absent | Present |
| WBC casts | ABSENT | Present (pathognomonic) |
| Systemic signs | Absent | Present |
| Renal impairment | No | Possible in severe/complicated cases |
| Treatment duration | 3-7 days | 7-14 days |
Recent Evidence Note
High-Yield Summary for Step 1
- Most common organism: E. coli (gram-negative, enteric bacillus, P pili virulence factor)
- Most common route: Ascending infection (lower UTI → VUR → upper tract)
- Classic presentation: Fever + CVA tenderness + dysuria/pyuria
- Key urinalysis finding: WBC casts (= renal tubular origin = upper tract)
- Histology: Neutrophilic tubulitis + interstitial suppuration + spared glomeruli
- Major complications: Papillary necrosis (diabetics/SCD/obstruction), pyonephrosis, perinephric abscess
- First-line treatment: Fluoroquinolones (outpatient); aminoglycosides (empiric inpatient)
- Do NOT use: Nitrofurantoin for upper tract infection
Acute interstatil nephritis
"acute interstitial nephritis"[MeSH Terms]
Acute Interstitial Nephritis (AIN) - Step 1 Review
Definition
- Comprehensive Clinical Nephrology, 7th Ed., p. 890
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 862
Causes / Etiology
1. Drug-Induced AIN (Most Common ~70-90%)
| Drug Class | Key Examples |
|---|---|
| Beta-lactam antibiotics | Methicillin (classic/prototype), ampicillin, amoxicillin, cephalosporins |
| Sulfonamides | First class ever reported to cause AIN |
| Fluoroquinolones | Ciprofloxacin, levofloxacin |
| Rifampin | |
| NSAIDs | Special: can co-present with minimal change disease + nephrotic syndrome |
| Proton pump inhibitors (PPIs) | Omeprazole, pantoprazole - often diagnosed months later |
| Diuretics | Thiazides, furosemide |
| Allopurinol | Associated with granulomatous AIN |
| Immune checkpoint inhibitors | Anti-PD-1, anti-PD-L1, anti-CTLA-4 (rising incidence) |
| Cimetidine |
2. Infection-Related AIN
- Historically: Streptococcus (scarlet fever), diphtheria
- Now: Legionella, Mycoplasma, EBV, CMV, leptospirosis, hantavirus
- Key distinction from pyelonephritis: relative absence of neutrophils and failure to isolate organism from kidney parenchyma - suggests immune-mediated mechanism, not direct invasion
3. Systemic / Autoimmune
- Sjogren syndrome - classic cause
- SLE (with immune complex deposition)
- Sarcoidosis - granulomatous AIN
- TINU syndrome (Tubulointerstitial Nephritis and Uveitis) - young women + bilateral uveitis
- IgG4-related disease
Pathogenesis
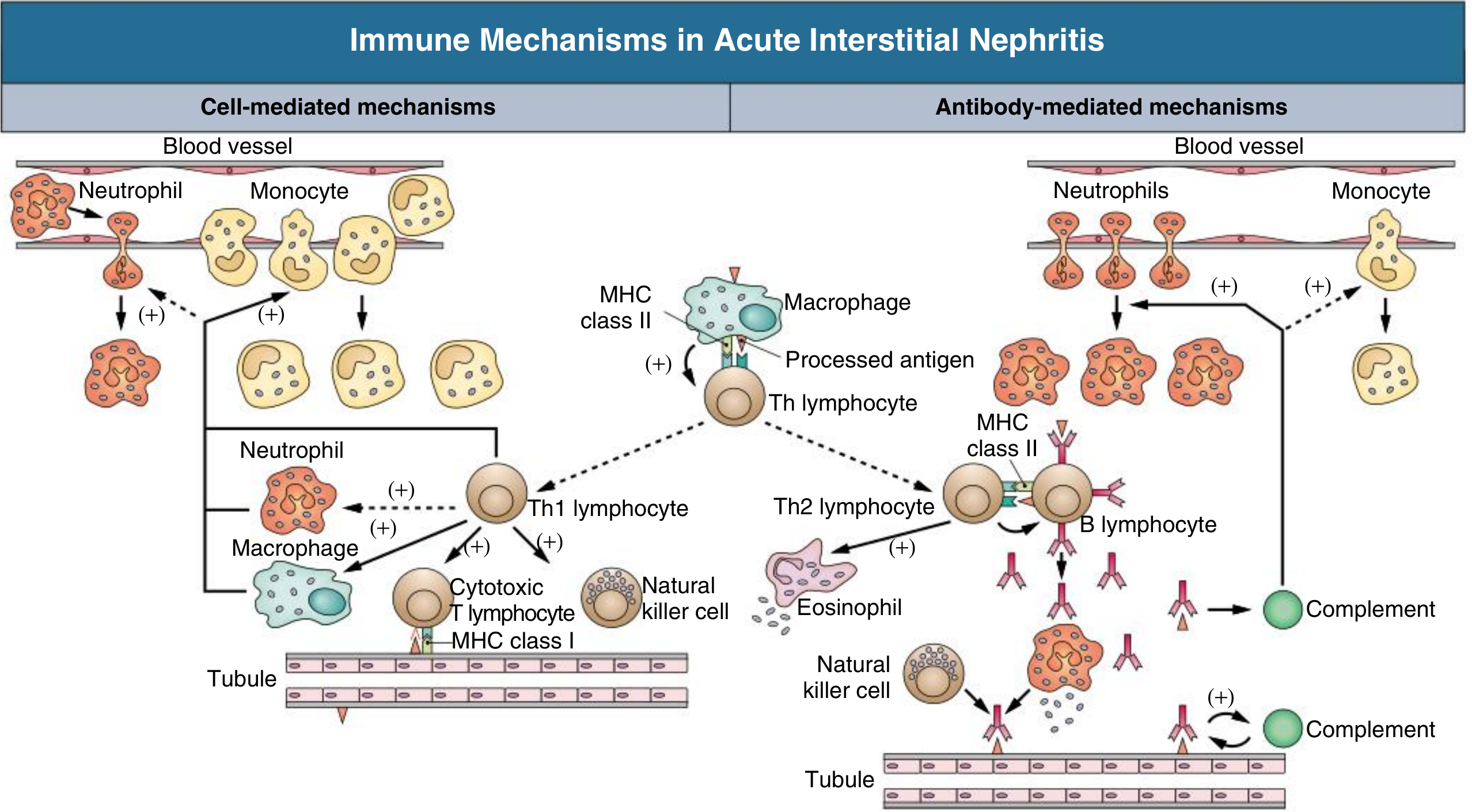
- Occurs in only a small fraction of people taking the drug
- Not dose-dependent
- Associated with systemic hypersensitivity signs (rash, eosinophilia, fever)
- Recurs on re-exposure to the same or related drug
- Responds to steroids
-
Type IV (cell-mediated / delayed-type) hypersensitivity - T cells, macrophages (dominant in most cases)
-
Type I (IgE-mediated) - seen with eosinophilia, elevated IgE, in some cases
-
Cytokine release (TGF-β) → interstitial fibrosis if injury persists
-
Comprehensive Clinical Nephrology, 7th Ed., p. 890
Clinical Presentation
Classic Triad (Step 1 favorite)
Fever + Maculopapular rash + Eosinophilia
Full Clinical Picture
| Feature | Details |
|---|---|
| Fever | Present in <50% of cases |
| Rash | Maculopapular, ~25% of cases |
| Peripheral eosinophilia | Variable; may be transient |
| AKI | Most common presentation - rising creatinine, often asymptomatic initially |
| Flank pain | ~33% - due to acute distension of kidney capsule |
| Oliguria | Present in severe cases; dialysis needed in ~1/3 |
| Hypertension / edema | Usually absent (distinguishes from glomerulonephritis) |
| DRESS syndrome | Drug Rash, Eosinophilia, Systemic Symptoms - occurs in up to 40% with certain drugs |
- Onset: 2-40 days after drug exposure (NSAIDs/PPIs: often months later)
- Goldman-Cecil Medicine, p. 1267
Laboratory Findings
| Test | Finding | Notes |
|---|---|---|
| Serum creatinine | Elevated (first and most sensitive finding) | May be significantly elevated before symptoms |
| Urinalysis | Pyuria, hematuria, mild proteinuria | |
| WBC casts | Present (leukocyte casts) | Key finding - indicates tubular origin |
| Eosinophiluria | >5% eosinophils in urine | Supportive but low sensitivity (31%) and specificity (68%) - NOT reliable to rule in or out |
| RBC casts | ABSENT | If present, suggests glomerulonephritis instead |
| Proteinuria | <1-3 g/day (non-nephrotic range) | Except NSAIDs (minimal change co-lesion → nephrotic range) |
| Peripheral eosinophilia | Variable | More with antibiotics; less with NSAIDs |
| FENa | Often >1% | Tubular dysfunction |
| Serum IgE | May be elevated |
Morphology / Pathology
- Kidneys normal or slightly enlarged (edema)
- Increased cortical echogenicity on ultrasound
-
Interstitial edema (displaces tubules apart)
-
Mononuclear infiltrate: predominantly T lymphocytes (CD4+) and macrophages; also eosinophils, plasma cells, mast cells
-
Tubulitis - lymphocyte infiltration into tubular epithelium (tubules infiltrated by WBCs)
-
Inflammation often starts at corticomedullary junction where drug concentration is highest
-
Glomeruli SPARED (until late disease)
-
Granulomas - seen with methicillin, thiazides, allopurinol, sarcoidosis
-
No immune deposits by immunofluorescence (usually) - distinguishes from lupus nephritis, membranoproliferative GN
-
Robbins, Cotran & Kumar, p. 863
Comparison Table: AIN vs. Other AKI Causes
| Feature | AIN | ATN | Glomerulonephritis |
|---|---|---|---|
| Fever/rash | May be present | Absent | Absent |
| Eosinophilia | Yes (variable) | No | No |
| RBC casts | ABSENT | Absent | PRESENT |
| WBC casts | PRESENT | Granular casts | Rare |
| Proteinuria | <3 g/day (mild) | Mild | Often heavy (nephrotic) |
| Hematuria | Mild | Absent | Prominent |
| Biopsy | Interstitial infiltrate | Tubular necrosis | Glomerular changes |
| FENa | >1% | >1% | <1% (usually) |
Diagnosis
-
Clinical suspicion + urinalysis + drug history
-
Eosinophiluria: supportive but not reliable (low PPV)
-
Renal biopsy: definitive diagnosis - indicated when:
- AKI unexplained by prerenal causes or ATN
- Diagnosis uncertain and tissue needed to guide therapy
- No improvement after stopping offending drug
-
Goldman-Cecil Medicine, p. 1269
Treatment
- Remove the offending drug / treat underlying cause - most important step
- Corticosteroids - prednisone 0.75-1 mg/kg/day x ~1 week then taper (8-12 weeks total); benefit is modest but may accelerate recovery if started within 7-14 days of stopping drug and before significant fibrosis
- Dialysis if severe AKI (~1/3 of patients need it)
- Mycophenolate mofetil - alternative for steroid-refractory or steroid-intolerant cases
- Most cases are reversible with early recognition and intervention
- Brenner and Rector's The Kidney, p. 3040
Special Associations for Step 1
| Drug / Condition | Special Association |
|---|---|
| Methicillin | Prototype drug; classic AIN description |
| NSAIDs | AIN + minimal change disease simultaneously → nephrotic syndrome |
| PPIs | Delayed onset (months); rising incidence; often missed |
| Allopurinol, thiazides | Granulomatous AIN |
| Checkpoint inhibitors | (anti-PD1, anti-CTLA4) - AIN occurs ~3% of the time |
| Sjogren syndrome | Interstitial nephritis + tubular dysfunction (RTA) |
| TINU syndrome | AIN + bilateral anterior uveitis in young women |
| Sarcoidosis | Granulomatous AIN |
High-Yield Step 1 Summary
- Definition: Immune-mediated inflammatory infiltrate in renal interstitium → AKI
- Most common cause: Drugs (antibiotics > NSAIDs > PPIs)
- Classic triad: Fever + rash + eosinophilia (present in only ~10-15% - rarely complete!)
- Key urine findings: WBC casts (leukocyte casts) + eosinophiluria; NO RBC casts
- Proteinuria: Mild (<3 g/day), UNLESS NSAIDs → minimal change co-lesion → nephrotic
- Biopsy: Interstitial lymphocytes/eosinophils + tubulitis + spared glomeruli + NO immune deposits (usually)
- Treatment: Stop drug + corticosteroids
- Prognosis: Usually reversible if caught early; chronic fibrosis if unrecognized
Interstatil renal inflammation
Renal interstitium
The Renal Interstitium
What Is the Renal Interstitium?
- Tietz Textbook of Laboratory Medicine, p. 1850
- Brenner and Rector's The Kidney, p. 117
Compartments of the Interstitium
1. Cortical Interstitium
- Very sparse in the normal kidney - only 7-9% of cortical volume (tubules are closely packed)
- Divided into:
- Peritubular interstitium: space between tubular basement membranes and peritubular capillaries
- Periarterial interstitium: loose connective tissue sheath around interlobular and arcuate arteries; contains lymphatics and nerve fibers
- The fenestrated portion of the peritubular capillary wall faces the narrow interstitial slit - this facilitates rapid reabsorption of fluid and solutes from the tubules into the bloodstream
2. Medullary Interstitium
-
Much larger volume fraction than the cortex (expands progressively from outer to inner medulla)
-
Contains lipid-laden interstitial cells arranged in a characteristic ladder-like pattern across the loops of Henle and vasa recta
-
The extracellular space is rich in glycosaminoglycans (GAGs), creating a gelatinous matrix
-
This gelatinous matrix contains osmolytes - osmotically active molecules that help stabilize the medullary osmotic gradient essential for the countercurrent concentration mechanism
-
The medullary interstitium is the functional core of urine concentration
-
Tietz Textbook of Laboratory Medicine, p. 1850
Cell Types of the Renal Interstitium
1. Fibroblasts (Most Abundant Cell)
- Also called stellate or sustentacular cells in the cortex
- Form a network spanning between capillaries and tubules via "attachment plaques" of actin fibers
- Primary producers of extracellular matrix (collagen, fibronectin, laminin)
- In the cortex, fibroblasts express ecto-5'-nucleotidase (5'NT) - a useful immunohistochemical marker
- Located in the inner stripe of the outer medulla and inner medulla
- Contain numerous lipid inclusions
- Produce prostaglandins (especially PGE₂) - important in regulating blood pressure and medullary blood flow
- Contain α-smooth muscle actin and vimentin filaments
- In inflammatory disease, fibroblasts become activated → transform into myofibroblasts
- Express α-smooth muscle actin (αSMA)
- Drive interstitial fibrosis and tubular atrophy → progressive CKD
- Pericytes (see below) are now recognized as the main progenitor of myofibroblasts
2. Pericytes
- Contractile cells wrapped around peritubular capillaries
- Most abundant in the inner stripe of the outer medulla around descending vasa recta
- Regulate capillary tone and microvascular blood flow
- In inflammation: pericytes detach from capillaries, proliferate, and differentiate into myofibroblasts - directly driving fibrosis and capillary rarefaction
3. Dendritic Cells
- Most common immune cell in the healthy interstitium
- Stellate shape with long cytoplasmic extensions
- Sentinel cells - present antigens to T cells during immune responses
- Distinguished from fibroblasts by: absence of actin bundles under the plasma membrane; organelles clustered near nucleus
4. Macrophages
- Mostly found in the periarterial interstitium in healthy kidney
- Not abundant in health; expand dramatically in injury
- In inflammation, release cytokines that amplify fibrosis and tubular damage
5. Lymphocytes and Granulocytes
- Rare in the healthy kidney
- Prominent in acute interstitial nephritis (lymphocytes + eosinophils) and pyelonephritis (neutrophils)
Extracellular Matrix (ECM) of the Interstitium
| Component | Details |
|---|---|
| Collagen fibrils | Types I, III, V, VI, VII, XV |
| Fibronectin | Cell adhesion, matrix organization |
| Laminin | Basement membrane component |
| Sulfated + nonsulfated GAGs | Ground substance; creates gelatinous matrix in medulla; stabilizes osmotic gradient |
| Interstitial fluid | Continuous with the ground substance |
- Brenner and Rector's The Kidney, p. 117
Functions of the Renal Interstitium
| Function | Mechanism |
|---|---|
| Structural support | Fibroblasts scaffold tubules and capillaries; ECM provides framework |
| Urine concentration | Medullary interstitium maintains the corticomedullary osmotic gradient via GAG matrix and lipid-laden cells |
| Solute/fluid exchange | Peritubular interstitium is the intermediate space for reabsorbed water and solutes moving from tubule lumen → capillary |
| Erythropoietin (EPO) production | Cortical fibroblasts (specifically peritubular fibroblasts) are the primary source of EPO - this is why CKD with interstitial fibrosis → loss of EPO-producing cells → normocytic anemia |
| Immune surveillance | Dendritic cells and macrophages monitor for pathogens and damaged cells |
| Prostaglandin synthesis | Medullary lipid-laden fibroblasts produce PGE₂ - vasodilatory, regulates blood pressure and medullary perfusion |
| Vitamin D activation | Peritubular cells contribute to 1α-hydroxylase activity |
The Interstitium in Disease
Interstitial Expansion = Bad Prognostic Sign
- Cellular infiltration: lymphocytes, macrophages, eosinophils (AIN, pyelonephritis)
- Edema: early AIN - kidneys enlarge due to interstitial fluid
- Interstitial fibrosis: myofibroblast activation → excess collagen deposition → tubular atrophy → nephron loss
Interstitial Fibrosis and Tubular Atrophy (IFTA)
- Final common pathway of most kidney diseases
- Driven by: TGF-β, PDGF, FGF-2 → fibroblast/myofibroblast proliferation
- Characterized by: collagen accumulation (types I-V, VII), fibronectin, laminin deposition
- Results in: capillary rarefaction, tubular dropout, glomerulosclerosis (eventually)
Specific Disease Correlates
| Disease | Interstitial Finding |
|---|---|
| Acute interstitial nephritis (AIN) | Edema + lymphocyte/eosinophil infiltrate + tubulitis; NO immune deposits |
| Pyelonephritis | Neutrophilic infiltrate + tubular abscesses; begins in medulla → affects concentration first |
| Chronic pyelonephritis | Coarse irregular scars, dilated calyces, patchy lymphocytic infiltrate |
| Sjogren syndrome | Lymphocytic interstitial nephritis + distal RTA |
| Analgesic nephropathy | Papillary necrosis + chronic interstitial fibrosis |
| CKD (any cause) | IFTA - correlates with GFR loss better than glomerular changes alone |
- Guyton and Hall Medical Physiology, p. 432
Key Step 1 Pearls
- Normal cortical interstitium = 7-9% by volume - mostly empty space between packed tubules
- EPO is made by cortical peritubular fibroblasts → interstitial fibrosis in CKD → anemia
- Medullary lipid-laden cells produce prostaglandins (PGE₂) and maintain the osmotic gradient
- Pyclocytes are pericytes under stress → detach → become myofibroblasts → fibrosis
- Dendritic cells are the dominant immune cell in the normal interstitium
- Pyelonephritis starts in the medulla → early loss of urine concentration ability
- Interstitial fibrosis (IFTA) is the best histologic correlate of GFR decline in most kidney diseases
- AIN: interstitial edema + lymphocyte infiltrate + tubulitis with spared glomeruli
Show in diagram
Detailed anatomical and cellular diagram of the renal interstitium for medical students. Show two main panels: LEFT PANEL - "Cortical Interstitium": Cross-section showing peritubular capillary (fenestrated), proximal tubule and distal tubule with basement membranes, narrow interstitial slit between tubule and capillary, stellate fibroblast with long cytoplasmic extensions labeled, dendritic cell with stellate shape, rare lymphocyte, extracellular matrix (collagen fibers, fibronectin), periarterial interstitium around artery with lymphatics and nerves. Label: "7-9% of cortical volume". Highlight fibroblast expressing ecto-5-nucleotidase (5'NT) and EPO production arrow from fibroblast. RIGHT PANEL - "Medullary Interstitium": Show loop of Henle, vasa recta (descending), lipid-laden interstitial cells arranged in ladder-like pattern spanning between loop of Henle and vasa recta, pericyte wrapping around capillary, GAG-rich gelatinous extracellular matrix labeled, osmotic gradient arrow showing increasing osmolality cortex to papilla (300 → 1200 mOsm). Label lipid-laden cells producing PGE2. BOTTOM PANEL - "Interstitium in Disease": Three boxes side by side: Box 1 "Normal" - sparse interstitium, fibroblast Box 2 "AIN" - edema, lymphocytes, eosinophils infiltrating, tubulitis, spared glomerulus Box 3 "Fibrosis/CKD" - myofibroblasts (activated fibroblasts with alpha-SMA), collagen deposition, tubular atrophy, capillary rarefaction, pericyte detachment arrow Color coding: tubules = pink, capillaries = red/blue, fibroblasts = orange, immune cells = purple, matrix = light yellow, lipid-laden cells = green. Clean medical textbook illustration style with clear labels.
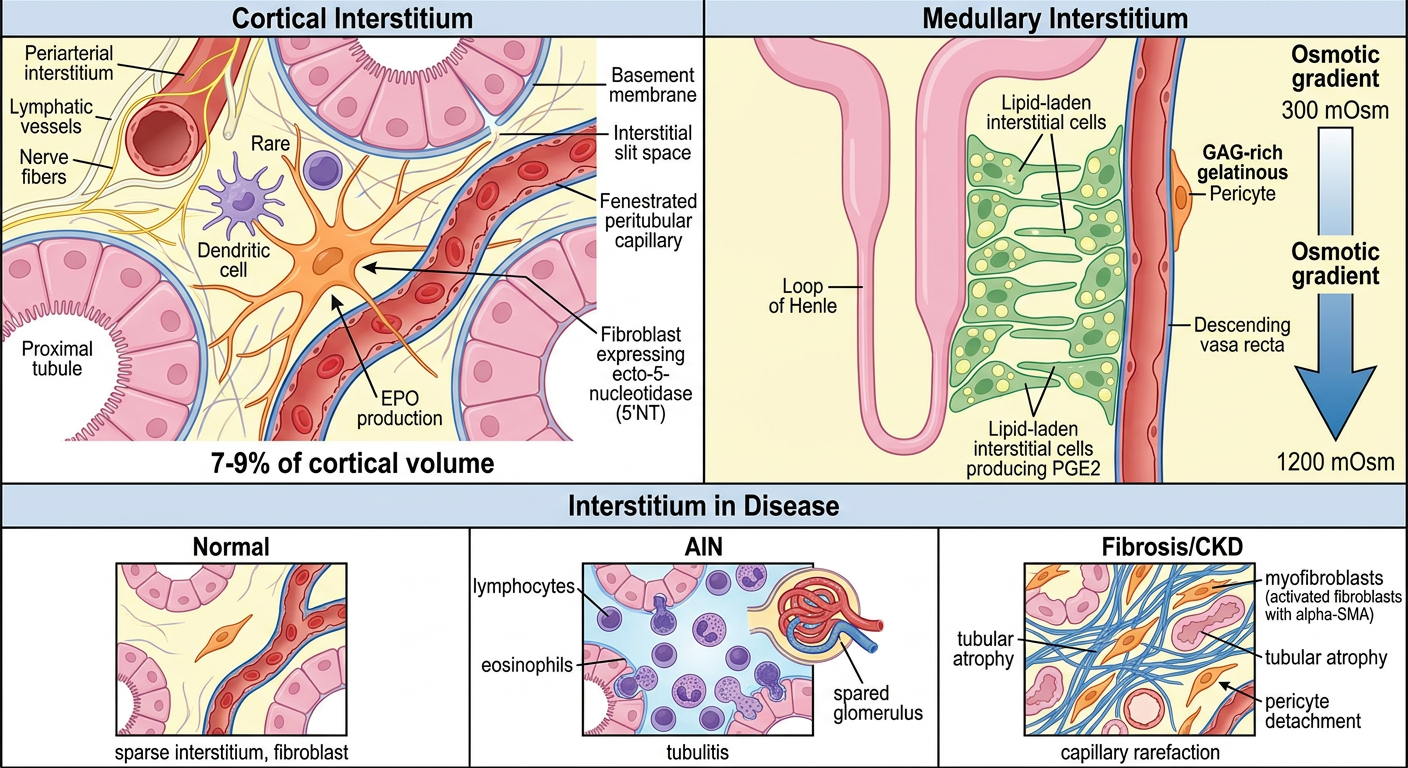
- The interstitium is very sparse (7-9% of cortical volume) - tubules are tightly packed
- Stellate fibroblasts (orange) with long extensions span between tubule basement membranes and peritubular capillaries - they produce EPO and ECM (collagen, fibronectin)
- Dendritic cells are the dominant immune cell in health
- Periarterial interstitium surrounds arteries with lymphatics and nerves
- Much larger volume fraction than cortex
- Lipid-laden interstitial cells (green) arranged in a ladder-like pattern crossing between loops of Henle and vasa recta - produce PGE₂ and help maintain the osmotic gradient (300 → 1200 mOsm)
- Rich GAG matrix creates gelatinous ground substance that stabilizes medullary osmolality
- Pericytes wrap around capillaries and regulate tone
- Normal: sparse, quiet fibroblasts
- AIN: interstitial edema + lymphocyte/eosinophil infiltrate + tubulitis; glomerulus spared
- CKD/Fibrosis: pericytes detach → become myofibroblasts (αSMA+) → collagen deposition → tubular atrophy + capillary rarefaction
Tell me about acute tubular necrosis every small concept for everything why it's occurring
Acute Tubular Necrosis (ATN) - Complete Deep Dive
What Is ATN? (Terminology First)
- Robbins & Kumar Basic Pathology, p. 519
- Frameworks for Internal Medicine, p. 454
Two Fundamental Types of ATN
| Type | Primary Cause | Segment Most Affected |
|---|---|---|
| Ischemic ATN | Decreased blood flow → hypoxia | S3 (straight) segment of proximal tubule + medullary thick ascending limb (mTAL) |
| Nephrotoxic ATN | Drugs, toxins, pigments | Primarily proximal tubule (S1, S2) - more uniform, more frank necrosis |
WHY THE PROXIMAL TUBULE AND mTAL?
The Proximal Tubule (especially S3 segment) is vulnerable because:
- High metabolic demand - it performs the most energy-demanding work (reabsorbs 60-70% of all filtered load). Requires massive amounts of ATP via oxidative phosphorylation. Cannot switch to anaerobic glycolysis effectively.
- Minimal mitochondrial reserve - when oxygen delivery drops, ATP falls rapidly → failure of Na⁺/K⁺-ATPase pumps → cell swelling and death
- High concentration of luminal toxins - as the filtrate is concentrated by water reabsorption, drugs and toxins accumulate in the tubular lumen at high concentrations, directly toxic to the epithelium
- High intracellular concentration of reabsorbed molecules - substances like organic acids, heavy metals, and aminoglycosides accumulate inside proximal tubular cells after uptake from the lumen
The Medullary Thick Ascending Limb (mTAL) is vulnerable because:
- Physiologically hypoxic environment - the outer medulla operates at the lowest PO₂ in the kidney due to the countercurrent exchange system (oxygen diffuses from descending to ascending vasa recta, "shunting" away from the medullary tubules)
- High metabolic demand - the mTAL actively reabsorbs NaCl (Na⁺/K⁺/2Cl⁻ cotransporter) and requires high ATP even under normal conditions
- No ability to extract more O₂ - already extracting nearly 100% of delivered oxygen; any reduction in delivery = immediate ischemia
- Robbins & Kumar Basic Pathology, p. 519
- Goldman-Cecil Medicine, p. 1265
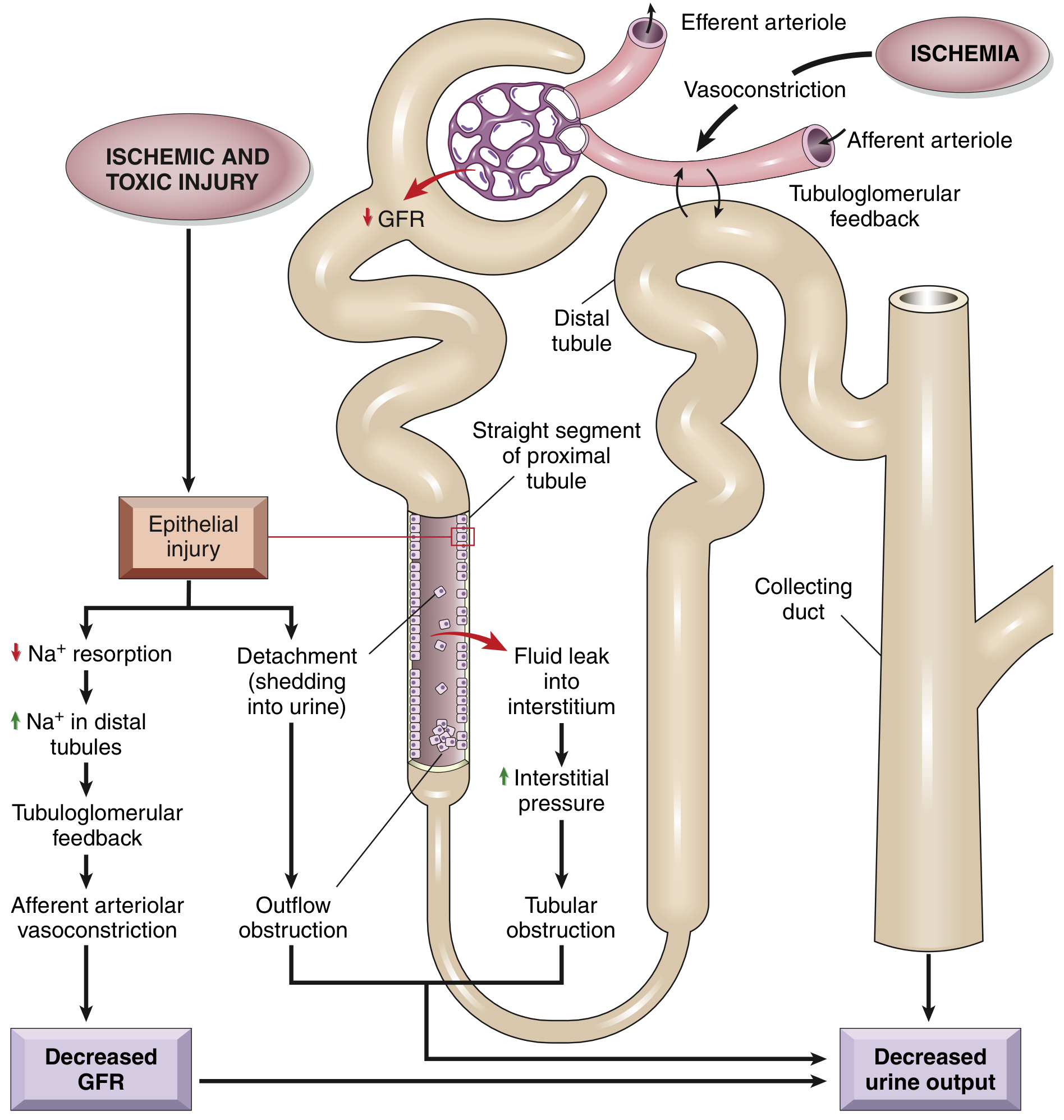
STEP 1: THE COMPLETE PATHOGENESIS - HOW DOES ATN CAUSE AKI?
Mechanism 1: Tubuloglomerular Feedback (TGF) → Afferent Vasoconstriction
- Ischemia/toxin → proximal tubule injury → failure of Na⁺ reabsorption
- More Na⁺ and Cl⁻ reaches the macula densa at the distal tubule
- Macula densa detects ↑ luminal NaCl → signals the juxtaglomerular apparatus
- Tubuloglomerular feedback activates → afferent arteriolar vasoconstriction
- ↓ Glomerular blood flow → ↓ GFR → further ischemia (a vicious cycle)
Mechanism 2: Vascular Factors - Endothelial Injury
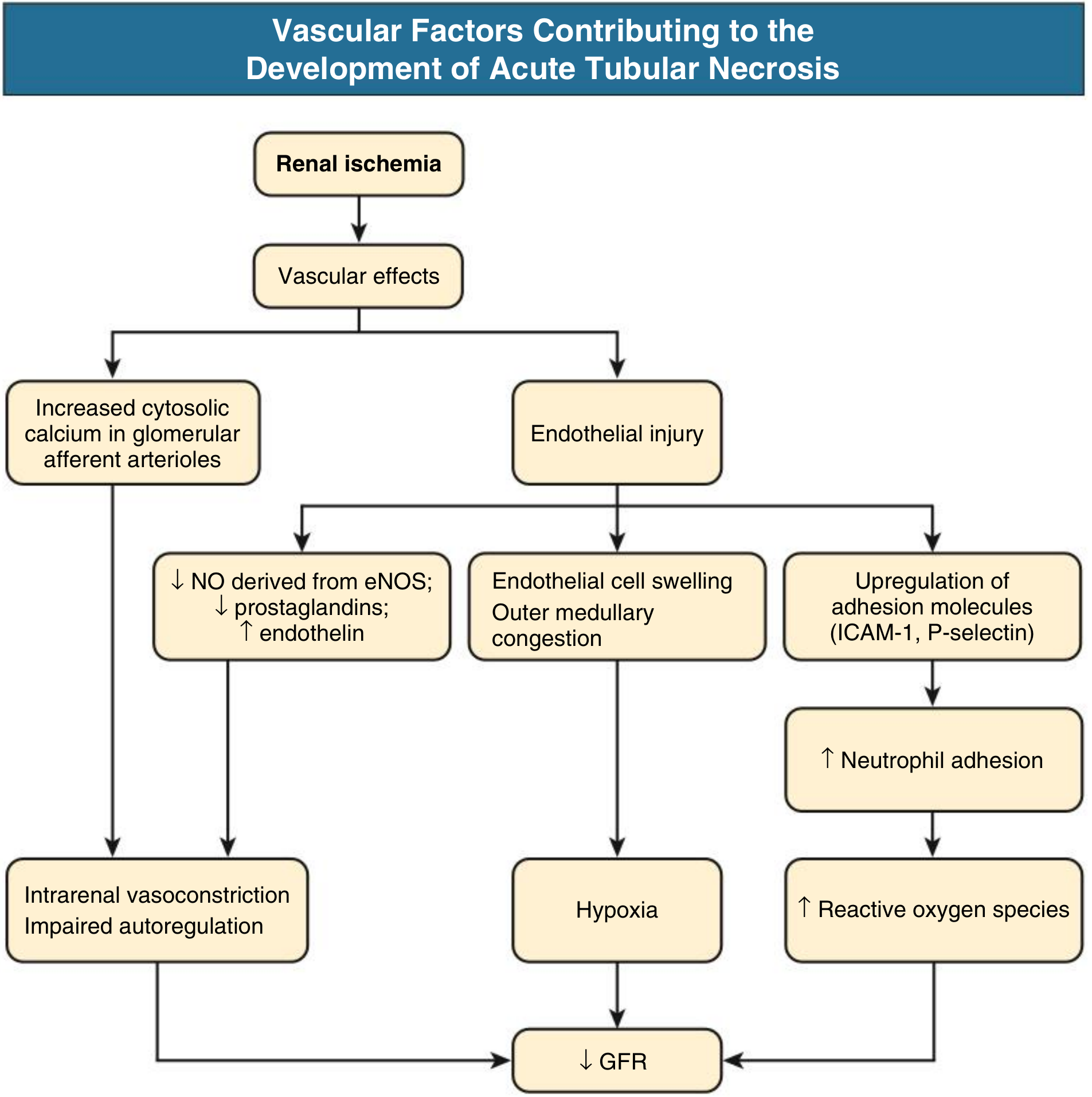
- ↓ eNOS → ↓ NO (normally vasodilatory) → vasoconstriction persists
- ↓ Prostaglandins (PGE₂, PGI₂) → loss of vasodilation
- ↑ Endothelin → potent vasoconstriction
- Endothelial cell swelling → outer medullary congestion → compresses tubules → hypoxia worsens
- Upregulation of ICAM-1 and P-selectin → neutrophil adhesion and infiltration → neutrophils release reactive oxygen species (ROS) and proteases → more tubular injury
Mechanism 3: Tubular Cell Injury - Loss of Polarity and Cast Formation
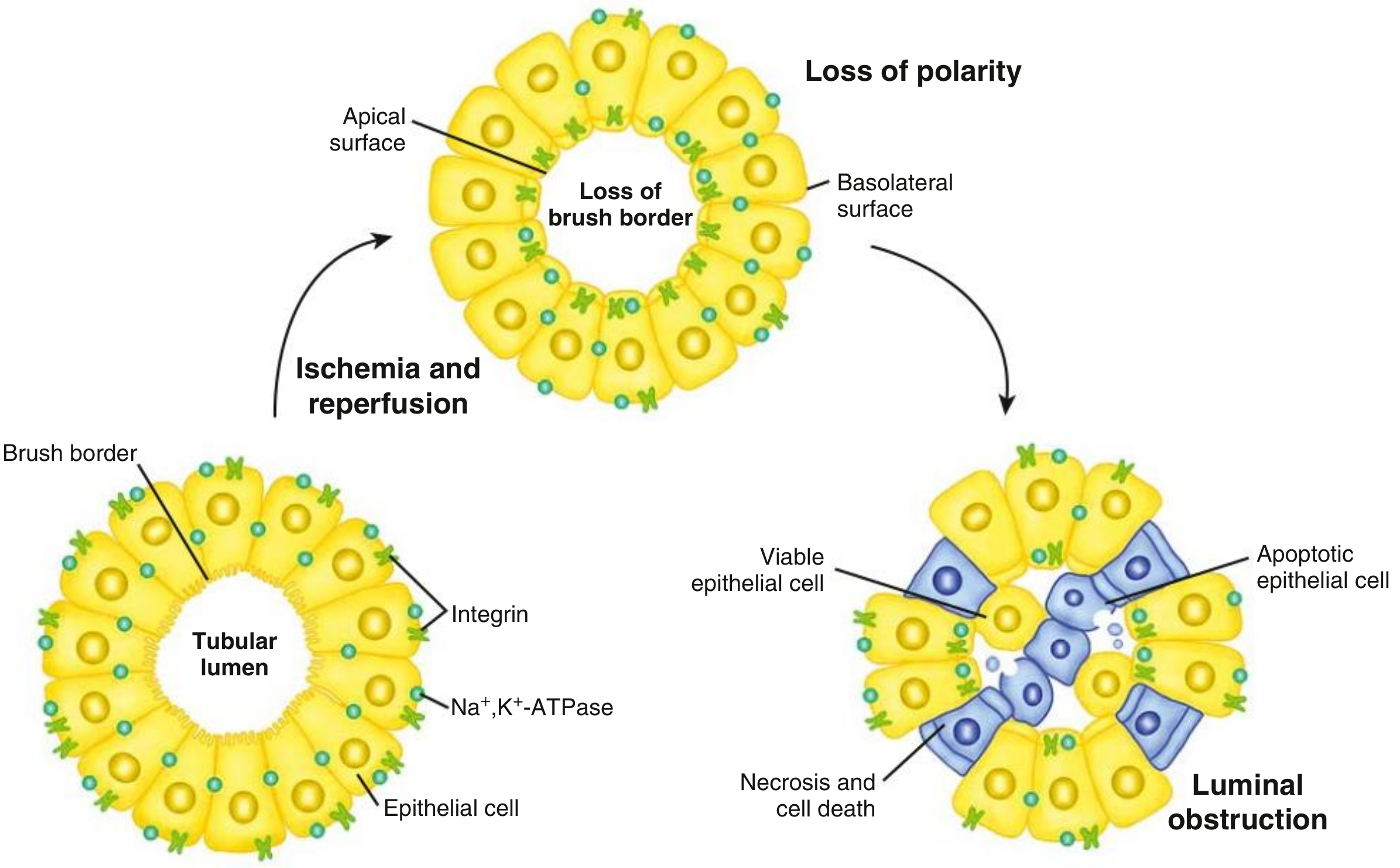
- Na⁺/K⁺-ATPase lives on the basolateral surface (pumps Na out into blood)
- Integrins and adhesion molecules keep the cell attached to the basement membrane
- ATP falls → Na⁺/K⁺-ATPase fails → intracellular Na⁺ and Ca²⁺ rise → cell swelling
- Loss of brush border (apical microvilli slough off) - first detectable change
- Loss of cell polarity - Na⁺/K⁺-ATPase redistributes from basolateral to apical surface → Na⁺ is now pumped backward into the lumen → more Na⁺ reaches the distal tubule (worsening TGF above)
- Integrins lose function → cells detach from the basement membrane
- Detached cells, cell debris, and Tamm-Horsfall protein aggregate in the tubular lumen → form "muddy brown" granular casts
- Casts obstruct tubular outflow → ↑ intratubular pressure → opposes glomerular filtration pressure → ↓ GFR
Mechanism 4: Backleak of Glomerular Filtrate
-
Glomerular filtrate leaks back through the denuded basement membrane into the interstitium
-
Filtrate is "lost" - never makes it to the urine
-
Creates interstitial edema → ↑ interstitial pressure → compresses capillaries → more ischemia
-
Results in oliguria despite ongoing filtration
-
Robbins & Kumar Basic Pathology, p. 520
-
Comprehensive Clinical Nephrology, 7th Ed., p. 990
CAUSES OF ATN - EVERY CATEGORY
A. Ischemic ATN (= prerenal → intrarenal continuum)
| Category | Examples |
|---|---|
| Hypovolemia / Hemorrhagic shock | Trauma, surgery, massive GI bleed, aortic surgery |
| Cardiogenic shock | MI, severe heart failure, cardiac arrest |
| Septic shock | Gram-negative bacteremia (most common cause of ATN in the ICU) |
| Distributive shock | Anaphylaxis, neurogenic shock |
| Obstetric emergencies | Placental abruption, eclampsia (→ cortical necrosis risk) |
| Large vessel disease | Renal artery thrombosis, embolism |
| Microvascular | Malignant hypertension, HUS/TTP, vasculitis |
B. Nephrotoxic ATN
| Agent | Mechanism / Notes |
|---|---|
| Aminoglycosides (gentamicin, tobramycin) | Accumulate in proximal tubule lysosomes → direct toxicity; non-oliguric ATN is characteristic; delayed onset (5-7 days) |
| Contrast agents (iodinated) | Direct tubular toxicity + vasoconstriction; risk ↑ with dehydration, DM, CKD, multiple myeloma, NSAIDs use |
| Cisplatin, carboplatin | DNA damage in proximal tubule cells |
| Amphotericin B | Pore-forming in tubular cell membranes + vasoconstriction |
| Vancomycin | Oxidative stress in tubular cells |
| NSAIDs | Block prostaglandin-mediated afferent dilation → ischemia (especially when dependent on PGs for GFR, e.g., CHF, CKD, elderly) |
| Heavy metals (mercury, lead, cisplatin, arsenic) | Direct cytotoxicity to proximal tubule |
| Ethylene glycol | Metabolized to oxalate → calcium oxalate crystals in tubules + direct toxicity |
| Carbon tetrachloride | Free radical-mediated injury |
| Agent | Source | Mechanism |
|---|---|---|
| Myoglobin | Rhabdomyolysis (crush injury, extreme exertion, statins, cocaine, hyperthermia) | Direct tubular toxicity + cast obstruction + vasoconstriction |
| Hemoglobin | Hemolytic crisis, mismatched blood transfusion, G6PD deficiency | Same as myoglobin |
| Uric acid | Tumor lysis syndrome | Crystal deposition and tubular obstruction |
| Light chains | Multiple myeloma | Cast nephropathy - obstructive + direct toxicity |
MORPHOLOGY - WHAT YOU SEE ON BIOPSY
Ischemic ATN:
- Patchy - lesions skip areas; not all tubules involved
- S3 segment of proximal tubule and mTAL are most affected
- Proximal tubule brush border loss - earliest change
- Cell blebbing, vacuolization, nuclear pyknosis
- Cells detach and slough → bare tubular basement membrane (key finding)
- Proteinaceous ("muddy brown") granular casts in distal tubules and collecting ducts
- Composed of: Tamm-Horsfall protein + plasma proteins + cell debris
- Basement membrane intact (allows regeneration!)
- Interstitial edema with mild neutrophil/lymphocyte infiltrate
- Glomeruli SPARED
Nephrotoxic ATN:
- More uniform, more proximal - concentrated in proximal tubule (S1/S2 segments)
- Frank necrosis more prominent than ischemic ATN (though still relatively sparse)
- Similar cast formation, basement membrane largely intact
- Robbins & Kumar Basic Pathology, p. 519-520
CLINICAL PHASES OF ATN
Phase 1: Initiation Phase (Hours to Days)
- The inciting event occurs (hypotension, toxin exposure)
- Oliguria begins
- BUN and creatinine start rising but are not yet markedly elevated (lag time)
- Histologic injury is occurring but clinical recognition may be delayed
- Key: If the trigger is removed NOW, tubular injury can be prevented
Phase 2: Maintenance Phase (Days to Weeks - typically 1-3 weeks)
- Established ATN - tubular cells are dead/dysfunctional
- Oliguria (urine output <400 mL/day) in ~50% - may be non-oliguric (especially aminoglycosides, contrast)
- Rising BUN and creatinine - azotemia
- Electrolyte complications:
- Hyperkalemia (↑K⁺ - tubules can't excrete K⁺; also released from injured cells)
- Metabolic acidosis (↓ NH₄⁺ excretion, ↓ H⁺ secretion)
- Hyperphosphatemia (↓ phosphate excretion)
- Hyponatremia (dilutional, if giving free water)
- Hypocalcemia (reciprocal to hyperphosphatemia)
- Fluid overload → pulmonary edema, hypertension
- Uremic symptoms: nausea, vomiting, lethargy, pericarditis (in severe/prolonged cases)
- Even if the cause is removed, GFR does not improve immediately - this is what distinguishes ATN from prerenal AKI
Phase 3: Recovery Phase (Weeks)
-
Tubular epithelial cells regenerate from surviving cells that dedifferentiate, proliferate, and re-cover the basement membrane
-
Surviving tubular cells (not stem cells) drive regeneration
-
Polyuria (diuretic phase) - urine output increases dramatically (can be 3-5 L/day)
- WHY: regenerating tubular cells cannot yet concentrate urine properly → pass large volumes of dilute urine
- Risk: patients can become severely dehydrated and hypokalemic during this phase
- Electrolyte losses (Na⁺, K⁺, Mg²⁺) can be life-threatening if not replaced
-
BUN and creatinine begin to fall
-
Function returns over weeks to months
-
If pre-existing CKD or severe prolonged injury: incomplete recovery → progression to CKD
-
M1 macrophages (pro-inflammatory) are replaced by M2 macrophages (reparative) during recovery
-
Robbins & Kumar Basic Pathology, p. 580
-
Comprehensive Clinical Nephrology, p. 959
LABORATORY FINDINGS & DIAGNOSIS
Urinalysis - The Most Important Test
| Finding | ATN | Prerenal AKI | AIN |
|---|---|---|---|
| "Muddy brown" granular casts | PRESENT - hallmark | Absent (hyaline casts only) | WBC casts |
| Tubular epithelial cell casts | Present | Absent | Absent |
| Proteinuria | Mild-moderate | Absent/trace | Mild |
| Hematuria | Absent | Absent | Mild |
| WBC casts | Absent | Absent | Present |
| RBC casts | ABSENT | Absent | Absent |

Fractional Excretion of Sodium (FENa)
| Result | Interpretation |
|---|---|
| FENa <1% | Prerenal AKI - tubules intact, avidly reabsorbing Na⁺ |
| FENa >2% | ATN - damaged tubules can't reabsorb Na⁺, Na⁺ is wasted |
- Tubular cells are dead/dysfunctional → Na⁺/K⁺-ATPase fails
- Na⁺ reabsorption is impaired → Na⁺ "spills" into the urine
- Also: Na⁺/K⁺-ATPase mislocalizes to apical surface → actively pumps Na⁺ INTO the lumen
- Contrast nephropathy (early)
- Myoglobinuria/hemoglobinuria
- Recent diuretic use (use FEUrea instead - >35-50% suggests ATN)
- ATN superimposed on prerenal disease
Other Labs
| Test | ATN Finding |
|---|---|
| Serum creatinine | ↑↑ (rises 0.5-1 mg/dL/day in oliguria) |
| BUN | ↑↑ (BUN:Cr ratio ~10-15 in ATN; >20 suggests prerenal) |
| K⁺ | ↑ (hyperkalemia) |
| HCO₃⁻ | ↓ (metabolic acidosis) |
| Phosphate | ↑ |
| Ca²⁺ | ↓ |
| Urine osmolality | ~300 mOsm/kg (isosthenuria) - tubules can't concentrate |
| Urine Na⁺ | >40 mEq/L (tubules can't reabsorb Na⁺) |
INFLAMMATION IN ATN (High-Yield Mechanism)
- Injured tubular cells release DAMPs (damage-associated molecular patterns)
- TLR2 and TLR4 are upregulated on tubular cells → recognize DAMPs → produce chemokines
- Complement system activated via the alternative pathway → TNF-α, IL-6, IL-1β, MCP-1 release
- Neutrophils adhere to endothelium via ICAM-1 → enter peritubular spaces → release proteases and ROS → amplify injury
- M1 macrophages infiltrate kidney → pro-inflammatory → worsen injury acutely
- CD4+ and CD8+ T cells contribute to injury
- Regulatory T cells help limit injury
-
M1 macrophages convert to M2 phenotype → support epithelial repair
-
Surviving tubular cells dedifferentiate, proliferate, migrate, and redifferentiate to cover denuded basement membrane
-
Comprehensive Clinical Nephrology, p. 959
SPECIAL TYPES OF ATN
Contrast-Induced Nephropathy (CIN)
- Develops 24-48 hours after contrast exposure
- Usually recovers within 7-10 days
- Prevention: IV hydration (0.9% saline or isotonic NaHCO₃) before procedure; stop NSAIDs/metformin; minimize contrast volume
- Risk factors: CKD, DM, dehydration, CHF, myeloma, concurrent nephrotoxins
Rhabdomyolysis-induced ATN (Pigment Nephropathy)
- Myoglobin is filtered → precipitates in tubules → cast obstruction
- Myoglobin is directly toxic (oxidative injury) + vasoconstricts
- Classic findings: elevated CK (often >10,000 U/L), dark/tea-colored urine, dipstick positive for blood, no RBCs on microscopy, elevated LDH, hyperkalemia (K⁺ released from necrotic muscle), hyperphosphatemia, hypocalcemia (Ca²⁺ deposits in damaged muscle)
- Treatment: aggressive IV fluid resuscitation (key!) to flush casts; urine alkalinization (prevents myoglobin precipitation); dialysis if needed
Aminoglycoside ATN
- Non-oliguric ATN - urine output is preserved (important distinction)
- Delayed onset: 5-7 days after starting drug
- Aminoglycosides accumulate in proximal tubule lysosomes → lysosome rupture → cell death
- Risk increased with: prolonged therapy, older age, CKD, volume depletion, concurrent loop diuretics
TREATMENT
- Remove/treat the cause - stop offending drug, restore perfusion
- Fluid management - correct hypovolemia; avoid overload; strict ins/outs
- Electrolyte management:
- Hyperkalemia: calcium gluconate (cardiac protection), insulin+dextrose, kayexalate, dialysis
- Metabolic acidosis: bicarbonate if severe
- Nutritional support - adequate calories to minimize catabolism
- Renal replacement therapy (dialysis) - indications: A-E-I-O-U:
- Acidosis (refractory)
- Electrolytes (hyperkalemia refractory to treatment)
- Ingestion (toxins)
- Overload (fluid refractory to diuretics)
- Uremia (encephalopathy, pericarditis, bleeding)
- During recovery phase: Replace electrolyte losses (Na⁺, K⁺) during polyuric phase; avoid diuretics which can worsen hypovolemia
PROGNOSIS AND RECOVERY
- Uncomplicated ATN with supportive care: complete recovery is common
- Time to recovery: days to weeks for mild injury; months for severe
- Non-oliguric ATN has a better prognosis than oliguric ATN
- Poor prognostic factors: older age, pre-existing CKD, multiorgan failure, prolonged oliguria
- Severe/prolonged injury → maladaptive repair → IFTA → CKD/ESRD
COMPLETE STEP 1 HIGH-YIELD SUMMARY
| Feature | ATN |
|---|---|
| Most common cause of AKI (inpatient) | Yes - most common intrinsic AKI |
| Two types | Ischemic (patchy, S3 + mTAL) and Nephrotoxic (uniform, proximal tubule) |
| Why S3 and mTAL? | High O₂ demand + can't do anaerobic glycolysis + medullary hypoxia |
| Key pathogenesis | Epithelial injury → ↓Na reabsorption → TGF → afferent vasoconstriction; + cast obstruction; + backleak; + endothelial injury |
| Hallmark urine finding | "Muddy brown" granular casts |
| FENa | >2% (tubules can't reabsorb Na⁺) |
| Urine osmolality | ~300 mOsm (isosthenuria) |
| Urine Na⁺ | >40 mEq/L |
| BUN:Cr ratio | ~10-15 (vs. >20 in prerenal) |
| Oliguric vs. non-oliguric | Both occur; aminoglycosides/contrast → non-oliguric |
| Recovery phase danger | Polyuria → dehydration + hypokalemia + hyponatremia |
| Histology | Patchy tubular cell loss, brush border loss, granular casts in distal tubules, intact basement membrane, spared glomeruli |
| Key difference from prerenal | Doesn't improve with fluids; FENa >2%; muddy brown casts; azotemia persists |
Tell
Answers previous question
What Was Covered in the ATN Answer:
-
Definition & Terminology - Why "necrosis" is a misnomer; modern term is Acute Tubular Injury (ATI)
-
Two Types - Ischemic ATN vs. Nephrotoxic ATN
-
WHY the Proximal Tubule and mTAL are vulnerable - High O₂ demand, can't do anaerobic glycolysis, medullary hypoxia
-
Complete Pathogenesis (4 mechanisms):
- Tubuloglomerular feedback → afferent vasoconstriction → ↓GFR
- Vascular/endothelial injury → ↓NO, ↑endothelin, neutrophil ROS
- Tubular cell polarity loss → cast formation → luminal obstruction
- Backleak of filtrate → oliguria
-
All Causes - Ischemic shock types, all nephrotoxic drugs, pigment nephropathy, contrast
-
Morphology - Patchy tubular necrosis, brush border loss, muddy brown casts, intact basement membrane, spared glomeruli
-
3 Clinical Phases - Initiation → Maintenance (oliguria, hyperkalemia, acidosis) → Recovery (polyuric phase with risk of hypokalemia/dehydration)
-
Lab Findings - Muddy brown casts, FENa >2%, isosthenuria, urine Na >40, BUN:Cr ~10-15
-
Inflammation in ATN - TLRs, complement, M1→M2 macrophage shift, neutrophil ICAM-1
-
Special Types - Contrast nephropathy, rhabdomyolysis, aminoglycoside ATN
-
Treatment - Fluids, electrolytes, AEIOU dialysis indications, recovery phase management
- The FENa calculation in detail
- The polyuric recovery phase
- Rhabdomyolysis specifically
- Comparing ATN vs. prerenal vs. AIN
- Treatment/dialysis indications
Didn't understand tell in complete detail
Don't tell in short like you did in pathogensis form complete sentences I don't understand this way
Acute Tubular Necrosis (ATN) - Complete Explanation in Full Sentences
Starting From the Very Beginning - What Is ATN?
- Robbins & Kumar Basic Pathology, p. 519
Why Do Tubular Cells Get Injured? The Two Root Causes
- Guyton and Hall Medical Physiology, p. 429
Why Are the Proximal Tubule (S3 Segment) and the Medullary Thick Ascending Limb (mTAL) the Most Vulnerable?
- Goldman-Cecil Medicine, p. 1265
- Comprehensive Clinical Nephrology, 7th Ed., p. 957
The Pathogenesis - How Does Tubular Cell Injury Actually Reduce GFR and Cause AKI?
First Mechanism: Loss of Na⁺ Reabsorption → Tubuloglomerular Feedback → Afferent Arteriolar Vasoconstriction
Second Mechanism: Endothelial Injury and Vascular Dysfunction
Third Mechanism: Loss of Tubular Cell Polarity → Cast Formation → Luminal Obstruction
Fourth Mechanism: Backleak of Glomerular Filtrate
- Robbins & Kumar Basic Pathology, p. 520
- Goldman-Cecil Medicine, p. 1266
The Causes of ATN - Explained
- Goldman-Cecil Medicine, p. 1265-1266
The Three Clinical Phases - The Full Story
- Comprehensive Clinical Nephrology, p. 959
- Guyton and Hall Medical Physiology, p. 429
What You Find in the Lab and Why
- Frameworks for Internal Medicine, p. 454
Treatment - Explained Logically
- Brenner and Rector's The Kidney
- Goldman-Cecil Medicine
The Bottom Line - One Paragraph Summary
Explain same like this overflow incontinence urgency incontinence stress incontinence
Urinary Incontinence - Complete Explanation in Full Sentences
Starting From the Very Beginning - How Does the Bladder Normally Work?
- Smith and Tanagho's General Urology, 19th Ed., p. 513
STRESS URINARY INCONTINENCE (SUI)
What Is It?
Why Does the Outlet Fail?
Who Gets Stress Incontinence?
Treatment of Stress Incontinence
- Smith and Tanagho's General Urology, 19th Ed., p. 610-615
URGENCY URINARY INCONTINENCE (UUI)
What Is It?
Why Does the Detrusor Become Overactive?
Treatment of Urgency Incontinence
- Smith and Tanagho's General Urology, 19th Ed., p. 717-754
OVERFLOW INCONTINENCE
What Is It?
Why Does the Bladder Not Empty?
How Do You Diagnose Overflow Incontinence?
Treatment of Overflow Incontinence
- Smith and Tanagho's General Urology, 19th Ed., p. 792-812
Side-by-Side Comparison of All Three Types
Explain renal cysts disorder
Explain again
Renal Cystic Disorders - Complete Explanation in Full Sentences
Starting From the Very Beginning - What Is a Renal Cyst?
- Goldman-Cecil Medicine, p. 1294
The Big Picture - Classification of Renal Cystic Disorders
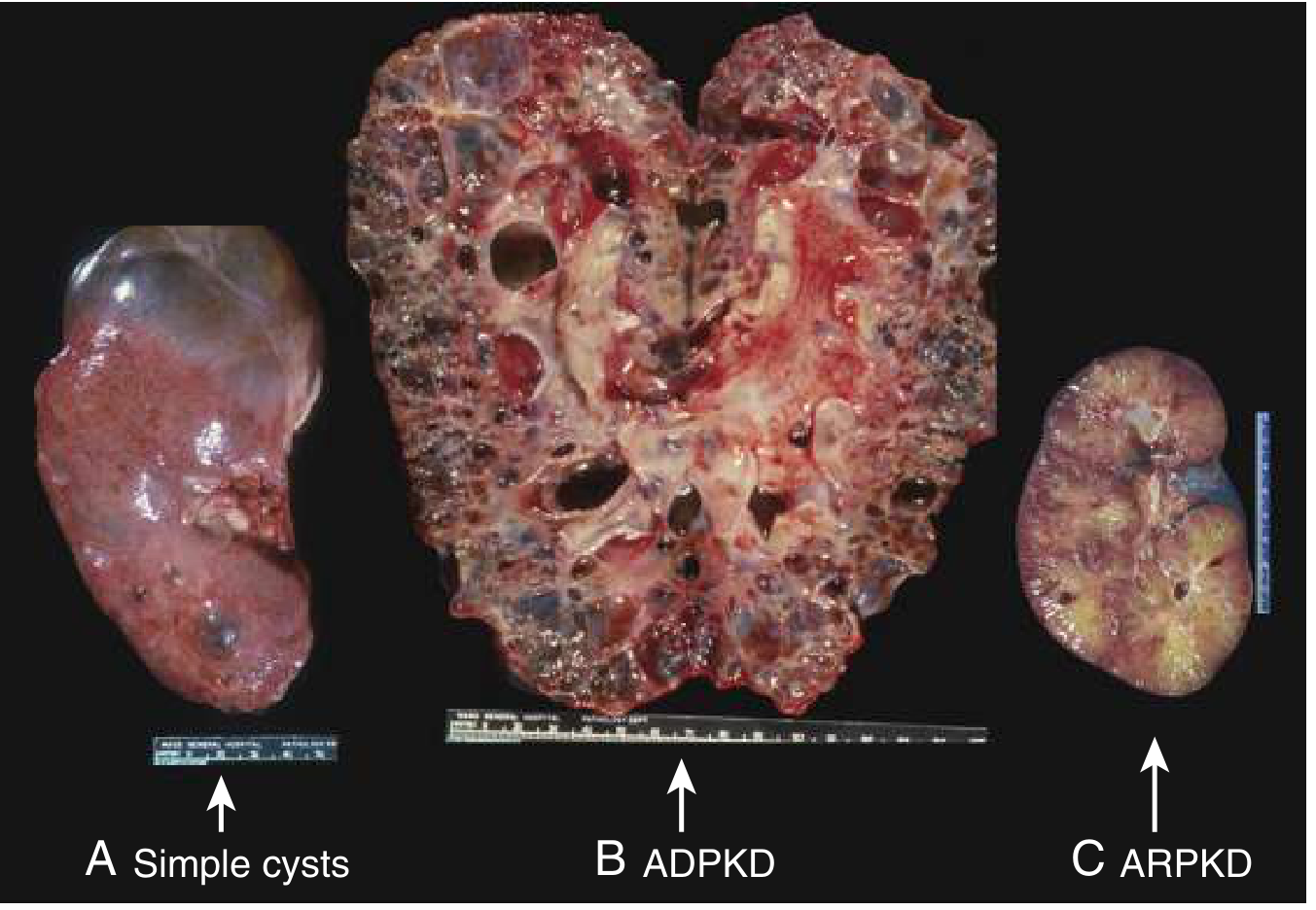
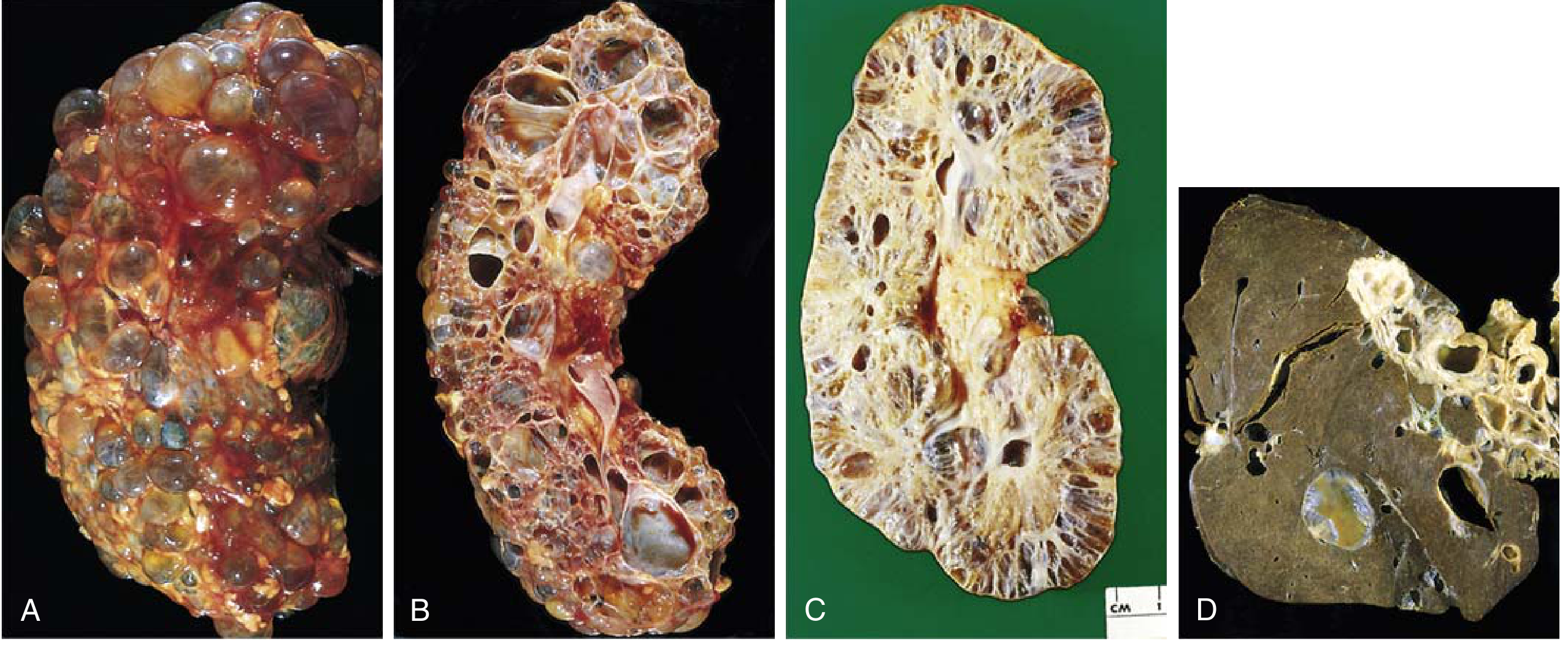
SIMPLE RENAL CYSTS
AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
What Is It and How Common Is It?
The Genetic Cause - PKD1 and PKD2
Why Do PKD1 Mutations Cause More Severe Disease Than PKD2?
How Do Cysts Destroy Kidney Function?
Clinical Features of ADPKD
Extra-Renal Manifestations - ADPKD Is a Systemic Disease
Diagnosis of ADPKD
Treatment of ADPKD
- Goldman-Cecil Medicine, p. 1295-1300
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE (ARPKD)
What Is It and How Is It Different From ADPKD?
How Does ARPKD Present?
The Liver Connection - Congenital Hepatic Fibrosis
- Robbins & Kumar Basic Pathology, p. 516
- Goldman-Cecil Medicine, p. 1300
NEPHRONOPHTHISIS (NPHP)
MEDULLARY SPONGE KIDNEY
ACQUIRED CYSTIC KIDNEY DISEASE
Quick Comparison - All the Key Differences
- Goldman-Cecil Medicine, p. 1294-1305
- The Developing Human - Clinically Oriented Embryology, p. 699
Renal cysts disorder
-
What a renal cyst is - how a tubular segment balloons out and fills with fluid
-
Simple renal cysts - benign, present in 50% of people over 40, Bosniak classification
-
ADPKD (Autosomal Dominant PKD)
- Most common hereditary kidney disease (1:400-1000)
- PKD1 (chr 16, 80%) vs PKD2 (chr 4, 15%)
- Role of primary cilium - polycystin proteins detect flow
- Why cysts form: loss of calcium signaling → cAMP rises → cell proliferation + chloride/fluid secretion
- Why PKD1 is worse than PKD2
- Clinical features: flank pain, hematuria, hypertension, UTIs, kidney stones
- Extrarenal features: liver cysts, intracranial berry aneurysms (5%), mitral valve prolapse, colonic diverticula
- Treatment: tolvaptan (V2 receptor blocker, only approved disease modifier) + ACE inhibitors
-
ARPKD (Autosomal Recessive PKD)
- PKHD1 gene → fibrocystin
- Presents at birth / in utero
- Tiny radially-oriented cysts from collecting duct
- Pulmonary hypoplasia → perinatal death in 25%
- Universally associated with congenital hepatic fibrosis → portal hypertension
- Potter sequence
-
Nephronophthisis
- Most common genetic cause of renal failure in children
- Normal/small kidneys, corticomedullary cysts + fibrosis
- Ciliopathy - associated with Joubert syndrome (molar tooth sign)
-
Medullary sponge kidney - dilated collecting ducts → nephrocalcinosis + stones, benign
-
Acquired cystic kidney disease - develops after years on dialysis → risk of renal cell carcinoma
Tell me in full explanation like you was telling yesterday
Tell me about renal cysts disorder in full paragraph every small explained
RENAL CYSTIC DISORDERS - Complete Explanation
PART 1 - What Is a Renal Cyst and Why Does One Form?
- Robbins & Kumar Basic Pathology, p. 522
- Goldman-Cecil Medicine, p. 1294-1295
PART 2 - SIMPLE RENAL CYSTS
- Robbins & Kumar Basic Pathology, p. 522
PART 3 - AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
What It Is and How Common It Is
The Genetic Cause - PKD1 and PKD2
How Cysts Grow and Destroy the Kidney
Clinical Features
Extrarenal Manifestations - ADPKD Is a Whole-Body Disease
Diagnosis
Treatment
- Robbins & Kumar Basic Pathology, p. 522-524
- Goldman-Cecil Medicine, p. 1295-1301
- Comprehensive Clinical Nephrology, p. 656-670
PART 4 - AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE (ARPKD)
What It Is
The Genetic Cause - PKHD1 and Fibrocystin
How ARPKD Looks and Why It Is Different Morphologically
Clinical Presentation - Severity Depends on Age at Diagnosis
The Liver in ARPKD - Congenital Hepatic Fibrosis
- Robbins & Kumar Basic Pathology, p. 524-525
- Comprehensive Clinical Nephrology, p. 668-669
PART 5 - NEPHRONOPHTHISIS (NPHP)
- Robbins & Kumar Basic Pathology, p. 525
PART 6 - MEDULLARY SPONGE KIDNEY (MSK)
PART 7 - ACQUIRED CYSTIC KIDNEY DISEASE (ACKD)
- Robbins & Kumar Basic Pathology, p. 522
PART 8 - VHL SYNDROME (Von Hippel-Lindau) - The Genetic Cyst/Tumor Syndrome
Summary Table - Key Differences at a Glance
- Robbins & Kumar Basic Pathology, pp. 522-526
- Goldman-Cecil Medicine, pp. 1294-1305
- Comprehensive Clinical Nephrology 7th Ed, pp. 656-671
Explain renal cell carcinoma
Make a presentation on renal cell carcinoma types and pathology
Purchase a credit top-up to continue using Orris