Krebs hensilit cycle
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed for a thorough answer. Here is the complete explanation:
The Krebs Cycle (Tricarboxylic Acid / Citric Acid Cycle)
The cycle has three names used interchangeably:
- Krebs cycle - named for Sir Hans Krebs who first described it
- Citric acid cycle - because citrate is the first product formed
- TCA cycle (tricarboxylic acid cycle) - because citrate and isocitrate carry three carboxyl groups
Location and Purpose
The TCA cycle takes place entirely in the mitochondrial matrix (except succinate dehydrogenase, which is embedded in the inner mitochondrial membrane). It is the final common pathway where catabolism of carbohydrates, fatty acids, and amino acids all converge. Its two main functions are:
- Complete oxidation of the acetyl group to CO2, capturing electrons as NADH and FADH2
- Providing carbon skeletons for biosynthetic reactions (gluconeogenesis, amino acid synthesis, heme synthesis)
(Lippincott Illustrated Reviews: Biochemistry, p. 332)
Entry Point: Pyruvate to Acetyl-CoA
Before entering the cycle, pyruvate (from glycolysis) is converted to acetyl-CoA by the pyruvate dehydrogenase complex (PDHC):
Pyruvate + CoA + NAD+ → Acetyl-CoA + CO2 + NADH
The PDHC requires five coenzymes: TPP (thiamine/B1), lipoic acid, FAD (riboflavin/B2), NAD+ (niacin/B3), and CoA (pantothenic acid/B5).
The 8 Steps of the TCA Cycle
Here is the complete cycle diagram from Guyton & Hall:
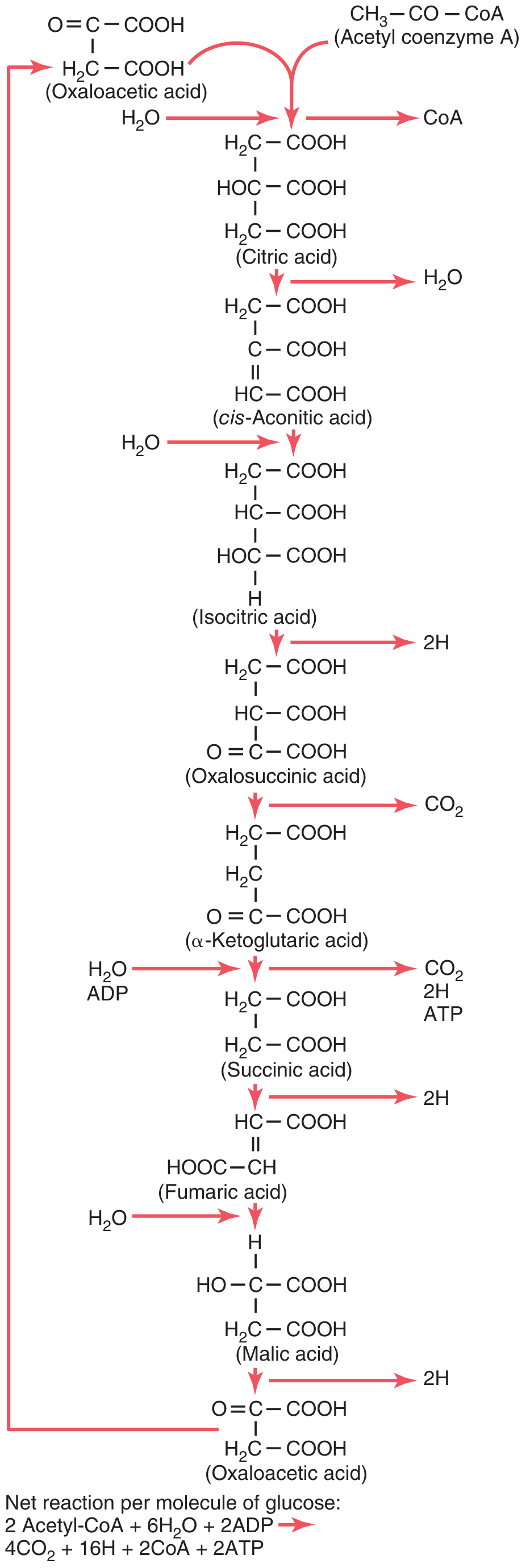
| Step | Reaction | Enzyme | Products |
|---|---|---|---|
| 1 | Oxaloacetate (4C) + Acetyl-CoA (2C) → Citrate (6C) | Citrate synthase | CoA released |
| 2 | Citrate → cis-Aconitate → Isocitrate | Aconitase | H2O removed then added |
| 3 | Isocitrate → α-Ketoglutarate | Isocitrate dehydrogenase | NADH + CO2 (1st CO2) |
| 4 | α-Ketoglutarate → Succinyl-CoA | α-Ketoglutarate dehydrogenase complex | NADH + CO2 (2nd CO2) |
| 5 | Succinyl-CoA → Succinate | Succinate thiokinase (succinyl-CoA synthetase) | GTP (substrate-level phosphorylation) |
| 6 | Succinate → Fumarate | Succinate dehydrogenase | FADH2 |
| 7 | Fumarate + H2O → Malate | Fumarase | - |
| 8 | Malate → Oxaloacetate | Malate dehydrogenase | NADH (3rd NADH) |
(Lippincott Illustrated Reviews: Biochemistry, pp. 339-344)
Energy Yield Per Acetyl-CoA (one turn)
| Coenzyme | Quantity | ATP equivalent |
|---|---|---|
| NADH | 3 | ~7.5 ATP (2.5 each via ETC) |
| FADH2 | 1 | ~1.5 ATP (1.5 via ETC) |
| GTP | 1 | 1 ATP (direct) |
| Total | ~10 ATP |
Since one glucose yields two acetyl-CoA, the TCA cycle contributes ~20 ATP per glucose (the majority of total ATP from aerobic glucose oxidation).
For the full glucose molecule:
- Net equation per glucose: 2 Acetyl-CoA + 6H2O + 2ADP → 4CO2 + 16H + 2CoA + 2ATP
(Guyton and Hall Textbook of Medical Physiology, p. [Fig 68.6])
Regulation of the TCA Cycle
Three key regulatory enzymes (all with large negative ΔG°):
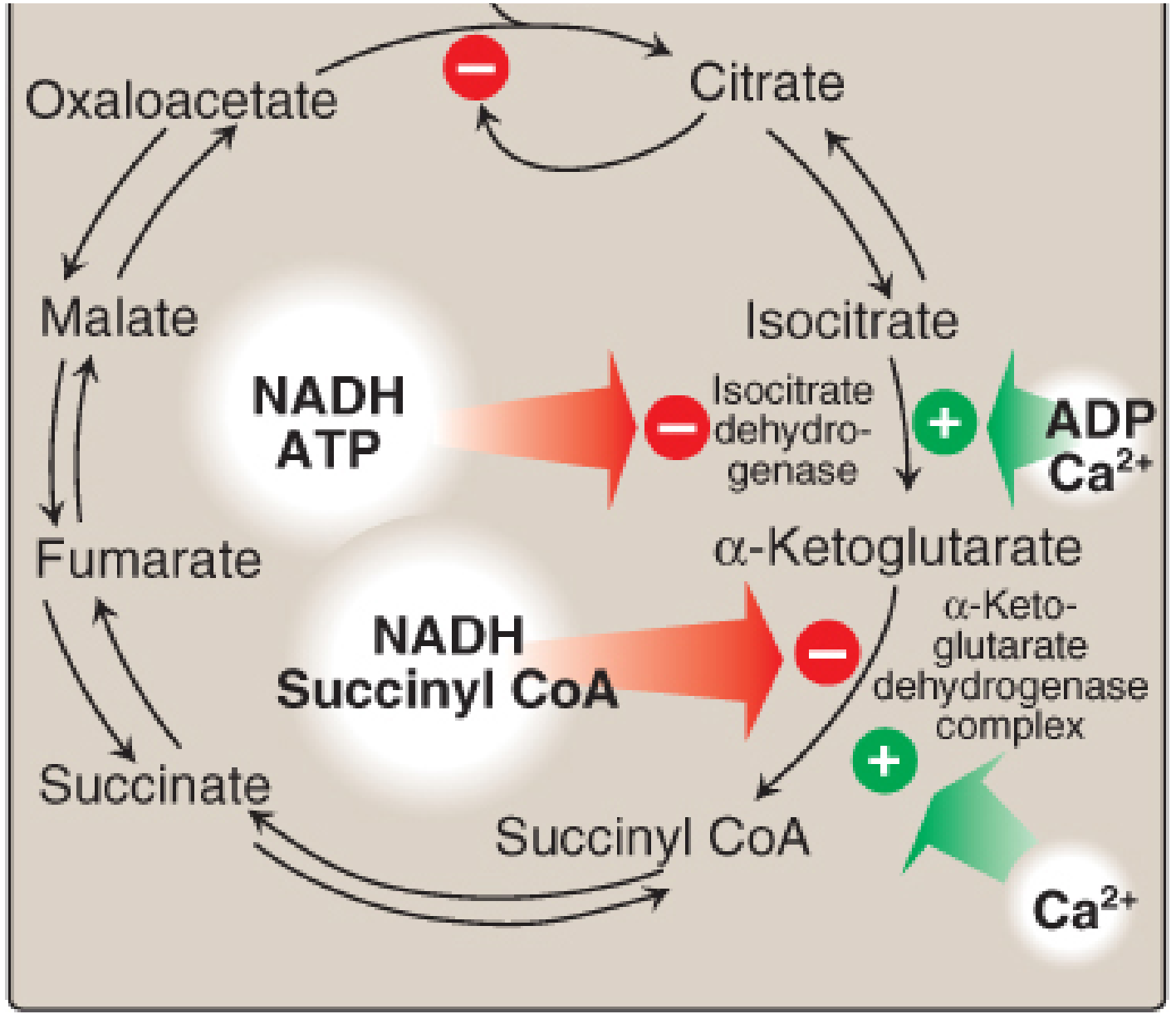
| Enzyme | Activated by | Inhibited by |
|---|---|---|
| Citrate synthase | Low acetyl-CoA, OAA | Citrate, NADH, succinyl-CoA, ATP |
| Isocitrate dehydrogenase | ADP, Ca2+ | ATP, NADH |
| α-Ketoglutarate dehydrogenase | Ca2+ | Succinyl-CoA, NADH |
The overall principle: when energy (ATP/NADH) is abundant, the cycle slows; when energy is low (ADP high), the cycle accelerates.
(Lippincott Illustrated Reviews: Biochemistry, p. 345-346)
Anaplerotic Reactions
These reactions "fill up" TCA cycle intermediates when they are drawn off for biosynthesis:
- Pyruvate carboxylase: Pyruvate + CO2 → Oxaloacetate (main anaplerotic reaction, requires biotin)
- Amino acid catabolism feeds in at multiple points (α-ketoglutarate from glutamate, succinyl-CoA from odd-chain fatty acids, fumarate from tyrosine/phenylalanine)
- OAA is also made from transamination of aspartate
Vitamins Required by the TCA Cycle
| Vitamin | Coenzyme Form | Where used |
|---|---|---|
| Thiamine (B1) | TPP | PDHC, α-KG dehydrogenase |
| Riboflavin (B2) | FAD | Succinate dehydrogenase, PDHC |
| Niacin (B3) | NAD+ | Isocitrate DH, α-KG DH, malate DH |
| Pantothenic acid (B5) | CoA | Acetyl-CoA, succinyl-CoA |
| Biotin | Biocytin | Pyruvate carboxylase (anaplerosis) |
Deficiency in thiamine (B1) is clinically important - it impairs both PDHC and α-KG dehydrogenase, causing lactic acidosis and neurological damage (Wernicke's encephalopathy).
(Basic Medical Biochemistry - A Clinical Approach, p. 827)
Key memory aid for intermediates (in order):
Oxaloacetate → Citrate → Isocitrate → α-Ketoglutarate → Succinyl-CoA → Succinate → Fumarate → Malate → OxaloacetateMnemonic: "Oh Citrate Is A Silly Stupid Fool's Mistake"
Urea cycle
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
I have everything needed for a thorough answer. Here it is:
The Urea Cycle (Krebs-Henseleit Cycle)
The urea cycle was first described in 1932 by Hans Krebs and his medical student Kurt Henseleit - making it the original "Krebs cycle" before the TCA cycle. It is the primary pathway for nitrogen excretion, converting toxic ammonia into the non-toxic, water-soluble compound urea for renal excretion.
Urea accounts for ~90% of the nitrogen-containing components of urine. (Lippincott Illustrated Reviews: Biochemistry, p. 712)
Where Does It Happen?
The urea cycle is a bicellular pathway - half in the mitochondrial matrix, half in the cytosol - and occurs only in the liver (hepatocytes).
- Steps 1-2: Mitochondrial matrix
- Steps 3-5: Cytosol
Nitrogen Sources Going Into Urea
Urea has two nitrogen atoms and one carbon atom:
- N-1 - from free ammonia (NH3), derived primarily from oxidative deamination of glutamate by glutamate dehydrogenase (GDH)
- N-2 - from aspartate (itself made by transamination of oxaloacetate)
- Carbon - from bicarbonate (HCO3-)
(Basic Medical Biochemistry, p. 1321)
The 5 Steps of the Urea Cycle
Here are the two cycle diagrams showing both the biochemistry and compartmentalization:
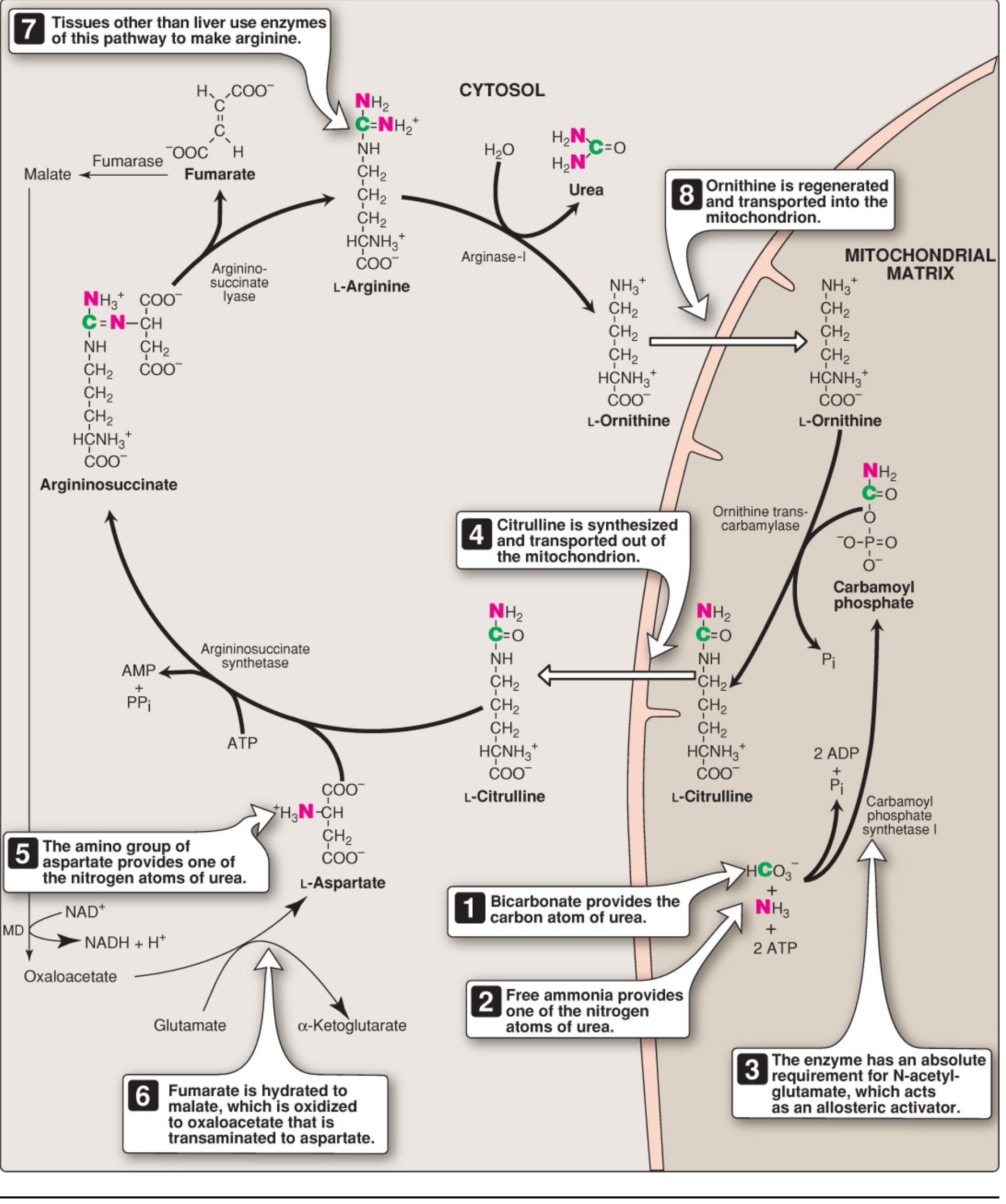
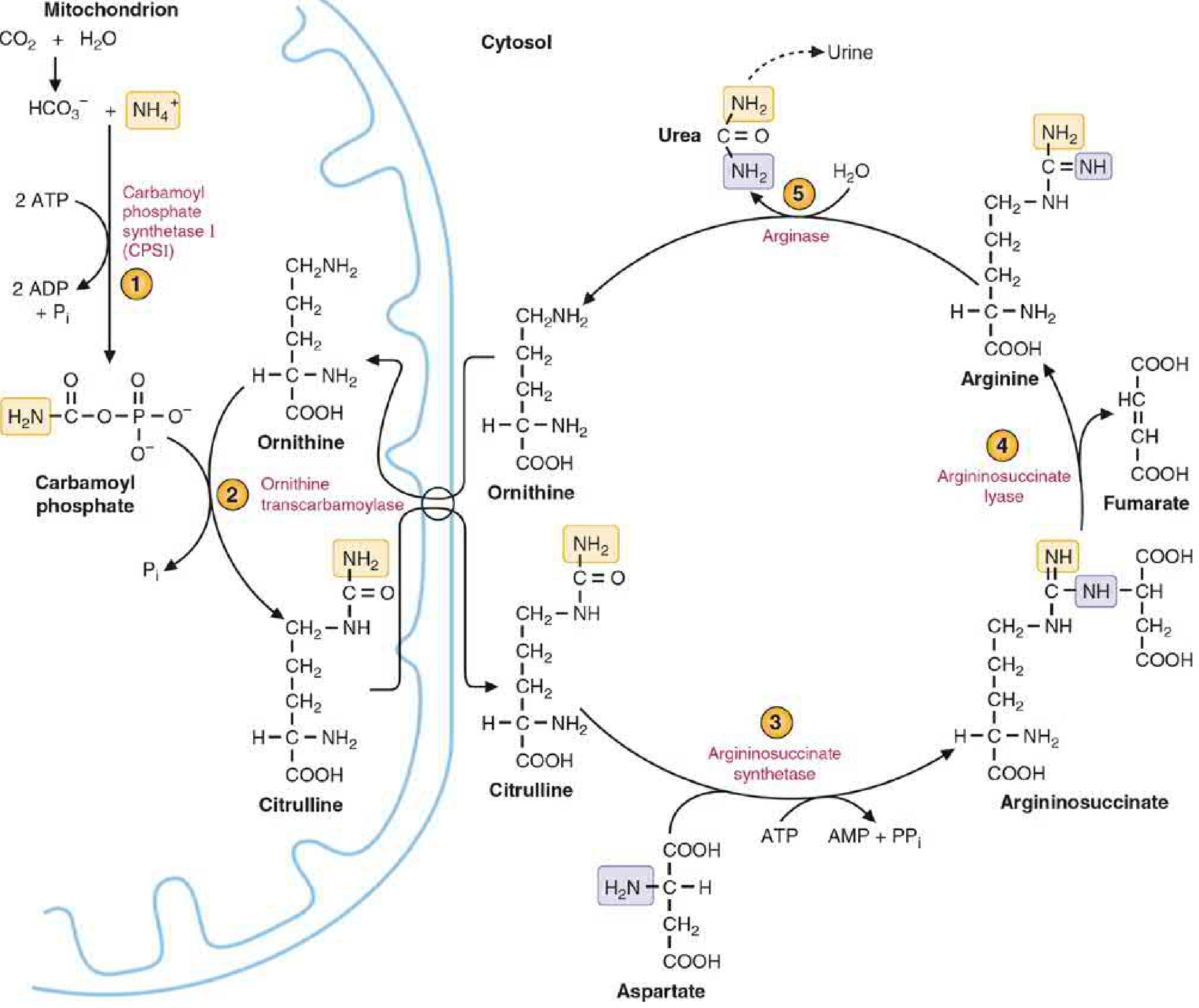
Step 1 - Carbamoyl Phosphate Formation (Mitochondria)
Enzyme: Carbamoyl phosphate synthetase I (CPS I)
NH3 + HCO3- + 2 ATP → Carbamoyl phosphate + 2 ADP + Pi
- Consumes 2 ATP - the most energy-expensive step
- Absolute requirement for N-acetylglutamate (NAG) as an allosteric activator
- NAG is synthesized from glutamate + acetyl-CoA by NAG synthase, activated by arginine
- CPS II (in cytosol) is a different enzyme used in pyrimidine synthesis - not part of the urea cycle
Step 2 - Citrulline Formation (Mitochondria)
Enzyme: Ornithine transcarbamylase (OTC)
Ornithine + Carbamoyl phosphate → Citrulline + Pi
- The carbamoyl group is transferred to ornithine
- Citrulline exits the mitochondria via an antiporter (exchanges with incoming ornithine)
- Ornithine is the carrier molecule analogous to oxaloacetate in the TCA cycle
Step 3 - Argininosuccinate Formation (Cytosol)
Enzyme: Argininosuccinate synthetase
Citrulline + Aspartate + ATP → Argininosuccinate + AMP + PPi
- Aspartate donates its amino group (the second nitrogen of urea)
- Consumes 1 ATP (cleaved to AMP + PPi = effectively 2 high-energy bonds)
- This is the rate-limiting step
Step 4 - Argininosuccinate Cleavage (Cytosol)
Enzyme: Argininosuccinate lyase
Argininosuccinate → Arginine + Fumarate
- Arginine is the immediate precursor of urea
- Fumarate is a key metabolic link - it enters the TCA cycle (or is hydrated to malate → transaminated to aspartate, replenishing the aspartate needed for step 3)
Step 5 - Urea Release (Cytosol)
Enzyme: Arginase-I (liver-specific)
Arginine + H2O → Urea + Ornithine
- Ornithine is regenerated and transported back into the mitochondria to restart the cycle
- Urea diffuses from the liver into blood and is excreted by the kidneys
- Arginase-I is virtually exclusive to the liver - this is why only the liver can complete urea synthesis
(Lippincott Illustrated Reviews: Biochemistry, pp. 712-715)
Overall Stoichiometry
Aspartate + NH3 + HCO3- + 3 ATP + H2O → Urea + Fumarate + 2 ADP + AMP + 2Pi + PPi
- 4 high-energy phosphate bonds consumed per urea molecule (2 ATP in step 1; 1 ATP→AMP in step 3)
- The reaction is energetically irreversible
Regulation
| Regulator | Mechanism | Effect |
|---|---|---|
| N-acetylglutamate (NAG) | Allosteric activator of CPS I | Activates whole cycle |
| Arginine | Activates NAG synthase | Increases NAG → activates CPS I |
| High protein diet / starvation | Increases expression of all urea cycle enzyme genes in liver | Upregulates cycle capacity |
| Substrate availability | Availability of NH3, HCO3-, aspartate | Controls flux |
The cycle is primarily regulated at CPS I through NAG. When protein catabolism is high (e.g. starvation, high-protein meal), more arginine and glutamate are produced, which stimulate NAG synthesis, which activates CPS I.
Link Between Urea Cycle and TCA Cycle
The fumarate produced in step 4 creates a direct link:
Fumarate (urea cycle)
↓ Fumarase
Malate
↓ Malate dehydrogenase
Oxaloacetate
↓ Transamination (AST)
Aspartate → back into urea cycle (step 3)
This is sometimes called the "urea cycle-TCA cycle bicycle" or the Krebs bicycle.
Urea Cycle Disorders (Hyperammonemia)
All urea cycle enzyme defects lead to hyperammonemia - accumulation of toxic ammonia. The most common is OTC deficiency (X-linked):
| Enzyme Defect | Accumulates | Key Features |
|---|---|---|
| CPS I deficiency | NH3 | No specific marker; hyperammonemia + encephalopathy |
| OTC deficiency (most common, X-linked) | Orotic acid, NH3 | Orotic aciduria distinguishes from CPS I deficiency |
| Argininosuccinate synthetase (Citrullinemia type I) | Citrulline | Elevated plasma citrulline |
| Argininosuccinate lyase (Argininosuccinic aciduria) | Argininosuccinate | Elevated argininosuccinate in urine/blood |
| Arginase deficiency | Arginine | Spastic diplegia, intellectual disability; low urea |
Why is ammonia toxic? It causes brain swelling by:
- Astrocytes convert NH3 + glutamate → glutamine (osmotic imbalance)
- Inhibits α-ketoglutarate dehydrogenase (disrupts TCA cycle)
- Inhibits glutaminase (further raises glutamine)
- Opens mitochondrial permeability transition pore → cell death
(Basic Medical Biochemistry, p. 1328)
Treatment of hyperammonemia:
- Low-protein diet
- Sodium benzoate / sodium phenylbutyrate (conjugate with amino acids, excreting nitrogen via kidney)
- Arginine supplementation (for defects after citrulline synthesis)
- Hemodialysis in acute crises
- Liver transplantation (definitive)
Memory Aid - The 5 Intermediates
Ornithine → Citrulline → Argininosuccinate → Arginine → Urea (back to Ornithine)Mnemonic: "Ordinarily Careless Crappers Are Also Frivolous About Urination" (Ornithine - Carbamoyl phosphate - Citrulline - Aspartate - Argininosuccinate - Fumarate - Arginine - Urea)
This is a shared conversation. Sign in to Orris to start your own chat.