PharmCidynamics
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I have everything I need for a comprehensive answer. Let me now compile the full pharmacodynamics guide.
Pharmacodynamics
Pharmacodynamics (PD) describes what a drug does to the body - the biochemical and physiological effects of drugs and their mechanisms of action. It answers: "How does this drug produce its effect?" It stands in contrast to pharmacokinetics (PK), which describes what the body does to the drug.
"Pharmacodynamics describes what the drug does to the body, particularly the relationship between drug concentration and pharmacologic effect." - Miller's Anesthesia, 10e
1. The Receptor Concept
Most drugs exert effects by interacting with specialized target macromolecules called receptors. The drug-receptor complex initiates alterations in biochemical or molecular activity through a process called signal transduction. - Lippincott Illustrated Reviews: Pharmacology
Key consequences of the receptor concept (Katzung's Basic and Clinical Pharmacology, 16e):
- Receptors determine quantitative relations - a receptor's affinity for a drug determines the concentration required for significant drug-receptor complex formation; the total number of receptors limits the maximal effect possible.
- Receptors confer selectivity - molecular size, shape, and electrical charge of a drug determine which receptor it binds.
- Receptors mediate agonism and antagonism - agonists activate receptors; antagonists bind but do not activate, blocking agonist access.
Most clinically relevant drug receptors are proteins - particularly regulatory proteins that mediate endogenous neurotransmitters, autacoids, and hormones. Other drug targets include enzymes (e.g., HMG-CoA reductase for statins; dihydrofolate reductase for methotrexate), transport proteins (e.g., Na⁺/K⁺ ATPase for digitalis glycosides; norepinephrine and serotonin transporters for antidepressants), and structural proteins (e.g., tubulin for colchicine).
2. Major Receptor Families
Four major families of receptors transduce drug signals (Lippincott Illustrated Reviews: Pharmacology):
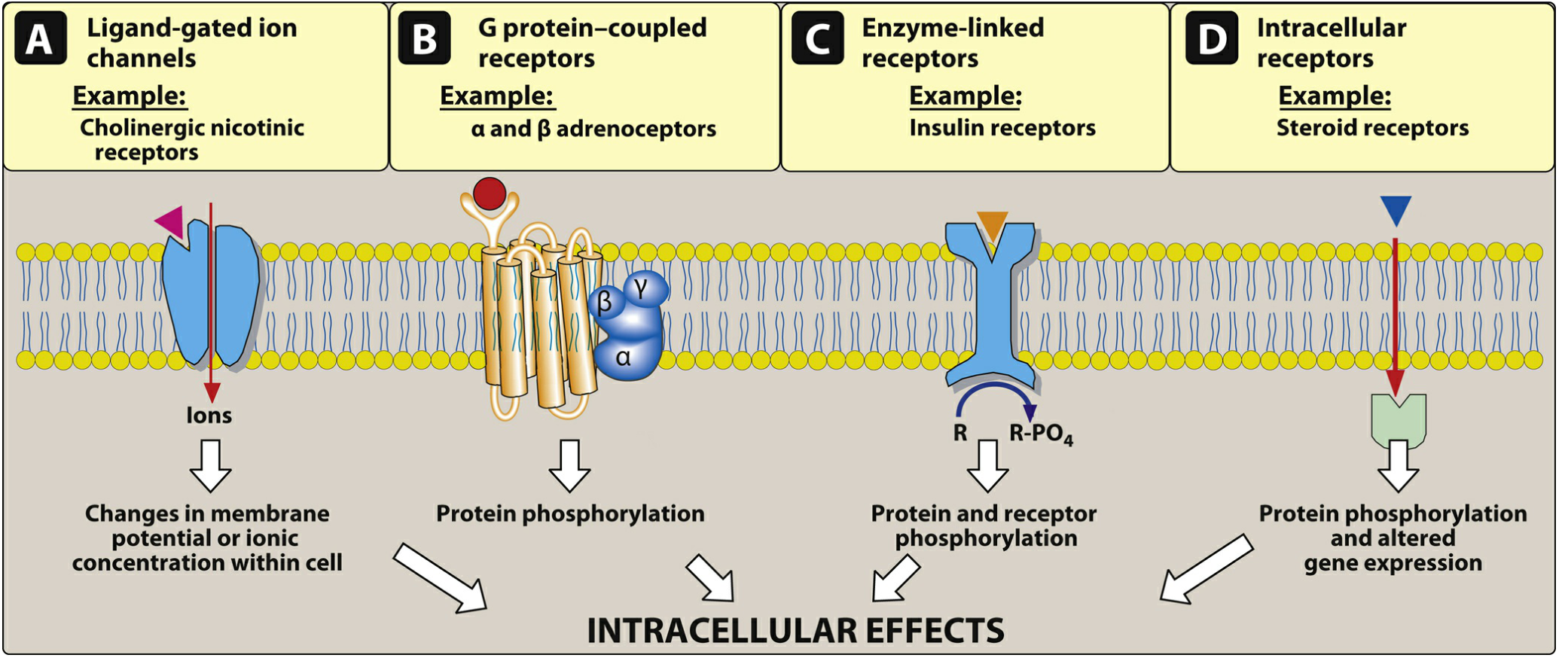
| Family | Location | Mechanism | Onset | Examples |
|---|---|---|---|---|
| Ligand-gated ion channels (ionotropic) | Cell membrane | Agonist binding opens ion pore - Na⁺/K⁺/Cl⁻ flow → membrane potential change | Milliseconds | Nicotinic ACh receptor, GABA-A receptor |
| G protein-coupled receptors (GPCRs, metabotropic) | Cell membrane | Agonist activates Gα subunit → GTP exchange → activates adenylyl cyclase, phospholipase C, or ion channels via second messengers (cAMP, IP₃, DAG) | Seconds-minutes | α/β adrenoceptors, muscarinic receptors, opioid receptors |
| Enzyme-linked receptors (receptor tyrosine kinases) | Cell membrane | Ligand binding activates intrinsic enzyme (usually tyrosine kinase) → protein/receptor phosphorylation | Minutes-hours | Insulin receptor, growth factor receptors |
| Intracellular receptors (nuclear receptors) | Cytoplasm/nucleus | Hydrophobic ligand crosses membrane → binds receptor → receptor-ligand complex enters nucleus → alters gene expression | Hours-days | Steroid receptors (glucocorticoids, estrogen, testosterone), thyroid hormone receptors |
Note on ligand-gated channels: Acetylcholine at nicotinic receptors opens a channel allowing Na⁺ influx and K⁺ efflux, generating an action potential. GABA-A receptor activation increases Cl⁻ influx → neuronal hyperpolarization (less likely to fire). Local anesthetics bind voltage-gated Na⁺ channels, inhibiting sodium influx and reducing neuronal conduction. - Lippincott Illustrated Reviews: Pharmacology
3. Receptor States and Intrinsic Activity
Receptors exist in at least two states - inactive (R) and active (R*) - in reversible equilibrium, usually favoring the inactive state. A drug's effect depends on which state it stabilizes. - Lippincott Illustrated Reviews: Pharmacology
| Drug Type | Intrinsic Activity | Emax | Effect |
|---|---|---|---|
| Full agonist | 1 (maximum) | = endogenous ligand | Fully activates receptor (e.g., phenylephrine at α₁ receptors) |
| Partial agonist | 0 < α < 1 | < full agonist | Partially activates receptor; can act as functional antagonist in presence of full agonist (e.g., buprenorphine at opioid receptors) |
| Antagonist | 0 | 0 | Binds but does not activate; blocks agonist access |
| Inverse agonist | Negative | Opposite of full agonist | Stabilizes inactive R state, producing effect opposite to agonist |
4. Dose-Response Relationships
As drug concentration increases, pharmacologic effect increases until all receptors are occupied. This produces a characteristic sigmoidal dose-response curve on a log-dose axis.
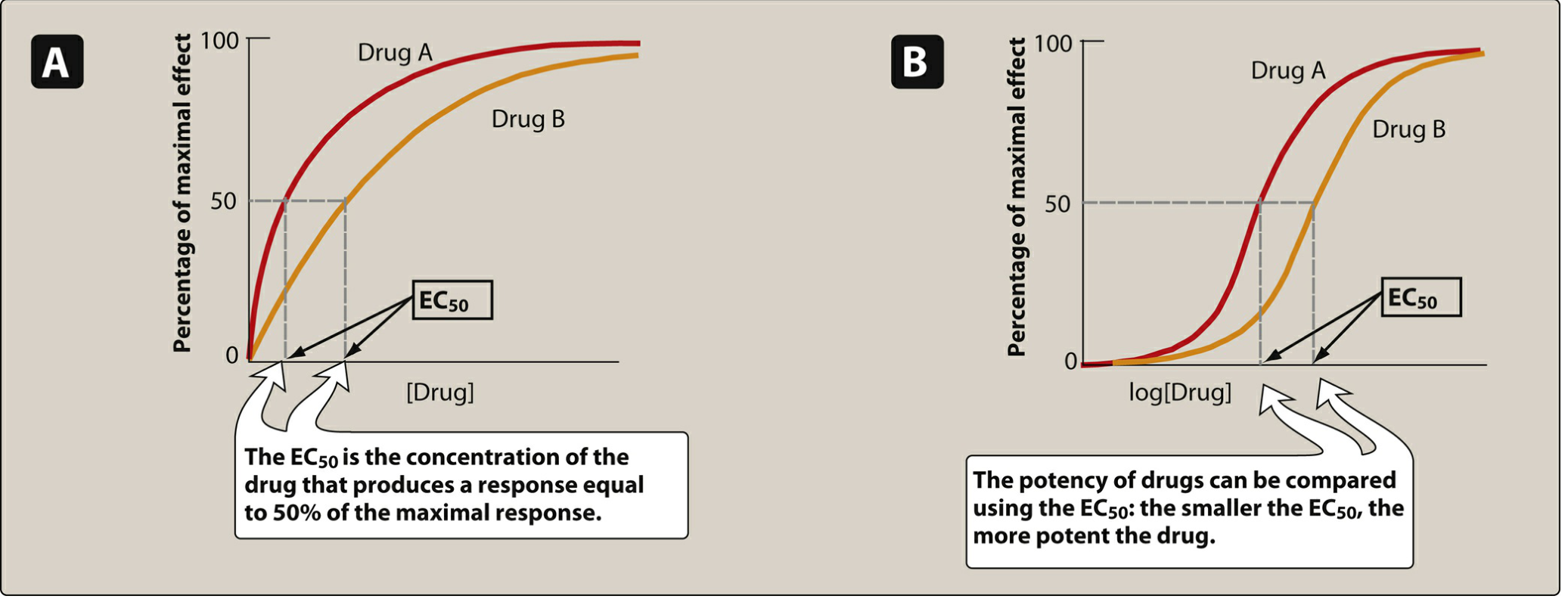
Potency
Potency is a measure of how much drug is required to produce an effect. It is expressed as EC₅₀ (the concentration producing 50% of maximum effect) or ED₅₀ (the dose producing 50% of maximum effect in a population). A smaller EC₅₀ = more potent drug.
Example: Candesartan (therapeutic dose 4-32 mg) is more potent than irbesartan (75-300 mg) - both are ARBs for hypertension. - Lippincott Illustrated Reviews: Pharmacology
Potency depends on: (1) receptor affinity (Kd) for the drug, and (2) efficiency of receptor-effector coupling.
Maximal Efficacy (Emax)
Maximal efficacy is the ceiling of the dose-response curve - the maximum effect a drug can produce regardless of dose. Drugs with the same target but different Emax have different clinical utility.
"The maximal efficacy of a drug is crucial for making clinical decisions when a large response is needed." - Katzung's Basic and Clinical Pharmacology, 16e
Important clinical distinction:
- A more potent drug is not necessarily more effective clinically.
- A partial agonist may be highly potent (low EC₅₀) but have lower Emax than a less potent full agonist.
5. Types of Antagonism
Competitive (Reversible) Antagonism
The antagonist competes with the agonist for the same binding site. The inhibition can be overcome by increasing agonist concentration - the dose-response curve shifts to the right (parallel shift) but Emax is preserved.
Schild equation: C'/C = 1 + [I]/K - where C' = agonist concentration needed in the presence of antagonist [I] with dissociation constant K. - Katzung's Basic and Clinical Pharmacology, 16e
Clinical example: Propranolol (competitive β-blocker) - at high sympathetic tone (exercise, stress), increased endogenous norepinephrine/epinephrine can overcome the block. The clinical effect of propranolol therefore varies with the degree of sympathetic activation.
Noncompetitive (Irreversible) Antagonism
The antagonist binds irreversibly (often covalently) or to an allosteric site. Increasing agonist concentration cannot overcome the block - Emax is reduced. Spare receptors can partially compensate if enough unoccupied receptors remain.
Allosteric Modulation
Some drugs bind to a site different from the endogenous ligand site. Benzodiazepines, for example, bind to GABA-A receptors at a site distinct from GABA, potentiating Cl⁻ influx without directly activating the channel - positive allosteric modulation.
6. Spare Receptors
Many receptor-effector systems have more receptors than are needed to produce a maximal response. These "spare receptors" (receptor reserve) mean that a maximal response may be produced by occupancy of only a fraction of total receptors. This has important consequences:
- In the presence of a low-dose irreversible antagonist, the agonist may still reach Emax (spare receptors allow it), but requires a higher dose.
- A higher-dose irreversible antagonist that eliminates all spare receptors will reduce Emax.
7. Quantal Dose-Response and Therapeutic Index
Quantal dose-response curves plot the frequency of an all-or-nothing response (e.g., "seizure" or "death") in a population against dose.
- ED₅₀: Dose effective in 50% of the population
- TD₅₀: Dose producing toxicity in 50% of the population
- LD₅₀: Lethal dose for 50% of the population
Therapeutic Index (TI)
$$TI = \frac{TD_{50}}{ED_{50}} \quad \text{or} \quad \frac{LD_{50}}{ED_{50}}$$
A high TI = wide margin between therapeutic and toxic dose (e.g., penicillins). A low TI = narrow margin requiring careful dosing (e.g., digoxin, warfarin, lithium, aminoglycosides).
The therapeutic window (or therapeutic range) is the plasma concentration range between the minimum effective concentration and the minimum toxic concentration.
8. Receptor Regulation
Receptors are not static targets - they change in number and sensitivity in response to drugs and disease:
- Downregulation: Chronic agonist exposure reduces receptor number or coupling efficiency. Example: chronic morphine use → opioid receptor downregulation → tolerance (need higher dose for same effect). - Lippincott Illustrated Reviews: Pharmacology
- Upregulation: Chronic antagonist use increases receptor density. Example: β-blockers upregulate β-adrenergic receptor density. Abrupt withdrawal → exaggerated catecholamine response (rebound tachycardia/hypertension). - Katzung's Basic and Clinical Pharmacology, 16e
- Tachyphylaxis/Desensitization: Rapid loss of response to repeated doses (e.g., biogenic amines).
- Sensitization: Increased receptor responsiveness with chronic antagonist use (e.g., clonidine withdrawal → hypertensive crisis due to upregulated α₂-adrenoceptors being flooded with endogenous norepinephrine).
9. Factors Modifying Pharmacodynamic Response
Age-Related Changes (Older Adults)
- Decreased sensitivity to β-adrenergic agonists and antagonists (but α₁ sensitivity unchanged)
- Increased sensitivity to benzodiazepines (altered GABA-A receptor function)
- Increased sensitivity to calcium channel blockers → greater BP and HR drop
- Decreased CNS dopamine → increased vulnerability to extrapyramidal effects from dopamine antagonists
- Decreased acetylcholine synthesis → increased anticholinergic neurotoxicity risk
- Enhanced warfarin effect at same plasma concentrations - ROSEN's Emergency Medicine
Disease-Specific Effects
Drug effects may differ between healthy individuals and patients with disease. Example: increasing dyspnea in a COPD patient on amiodarone could reflect drug toxicity, disease progression, or a new cardiopulmonary event. - Harrison's Principles of Internal Medicine, 22e
Receptor Up/Downregulation by Disease
Thyroid hormones increase β-adrenoceptor density in cardiac muscle - explaining tachycardia of thyrotoxicosis and the utility of propranolol for symptom control. - Katzung's Basic and Clinical Pharmacology, 16e
Pharmacogenetics and Precision Medicine
Genetic variants can affect pharmacodynamics directly (e.g., EGFR mutations in lung cancer that confer sensitivity to gefitinib, a tyrosine kinase inhibitor) or indirectly via altered signal transduction. Study of genetic factors determining drug response is pharmacogenetics; application to individualize therapy is precision medicine.
10. Time Course of Drug Action
Drug effect over time is not always simply explained by plasma concentration (PK). Reasons for delay or persistence of effect include:
- Slow target tissue uptake (e.g., cardiac glycosides distributing to myocardium)
- Slow accumulation of active metabolites
- Irreversible receptor binding - e.g., clopidogrel irreversibly inhibits P2Y12 receptors; plasma t½ ~6 hours but antiplatelet effect lasts 7-10 days (platelet lifespan). - Harrison's Principles of Internal Medicine, 22e
- Receptor changes distal to the primary target - e.g., antidepressant effects delayed due to downstream neuroplasticity
Quick Reference Summary
| Concept | Definition | Clinical Relevance |
|---|---|---|
| EC₅₀ | Concentration for 50% maximal effect | Measures potency |
| Emax | Maximum possible effect | Distinguishes full vs partial agonists |
| Therapeutic index | TD₅₀/ED₅₀ | Safety margin; guides dosing caution |
| Full agonist | Intrinsic activity = 1 | Produces maximal effect (e.g., morphine) |
| Partial agonist | 0 < α < 1 | Ceiling effect; can antagonize full agonists (e.g., buprenorphine) |
| Competitive antagonist | Reversible; shifts curve right | Effect overcome by more agonist (e.g., propranolol) |
| Noncompetitive antagonist | Irreversible; reduces Emax | Cannot be overcome by agonist |
| Downregulation | Decreased receptor number | Tolerance (e.g., opioids) |
| Upregulation | Increased receptor number | Withdrawal syndromes (e.g., β-blockers) |
Sources:
- Katzung's Basic and Clinical Pharmacology, 16th Edition, Chapter 2: Drug Receptors & Pharmacodynamics
- Lippincott Illustrated Reviews: Pharmacology - Chapters on signal transduction and dose-response
- Harrison's Principles of Internal Medicine, 22e - Principles of Pharmacodynamics
- ROSEN's Emergency Medicine - Pharmacodynamics in Older Adults
- Miller's Anesthesia, 10e - Pharmacodynamics Key Points
This is a shared conversation. Sign in to Orris to start your own chat.