Pleuroparenchymal fibroelastosis full details for md exams 10 marks answer
Reading File
Finding Sources
Finding Sources
Searching PubMed
pleuroparenchymal fibroelastosis
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive data from three authoritative textbooks (Murray & Nadel, Fishman's, Goldman-Cecil) plus recent PubMed reviews. Let me compile the full MD exam answer.
Pleuroparenchymal Fibroelastosis (PPFE)
A Rare Idiopathic Interstitial Pneumonia - Complete 10-Mark Answer
1. Definition and Historical Background
Pleuroparenchymal fibroelastosis (PPFE) is a rare, upper zone-predominant fibrotic lung disease characterized by visceral pleural thickening and subpleural parenchymal fibrosis with prominent elastosis.
- The term "PPFE" was first coined by Frankel in 2004, describing five patients with a previously uncategorized fibrotic pattern.
- It closely resembles Pulmonary Upper Lobe Fibrosis (PULF) described by Amitani et al. in 1992 - both are now accepted as the same entity.
- Formally recognized as an independent rare Idiopathic Interstitial Pneumonia (IIP) by the ATS/ERS consensus statement in 2013.
- Accounts for approximately 6-8% of all ILDs, though the true incidence is unknown due to lack of formal diagnostic criteria and frequent misdiagnosis.
2. Classification
PPFE falls under the Rare IIPs category in the ATS/ERS 2013 classification of Idiopathic Interstitial Pneumonias (alongside Idiopathic LIP).
It can be:
| Type | Description |
|---|---|
| Idiopathic PPFE | No identifiable cause; most common form |
| Secondary PPFE | Associated with specific conditions (see Associations below) |
3. Etiology and Associated Conditions
The etiology is largely unknown. Secondary causes and associations include:
Transplantation:
- Hematopoietic stem cell transplantation (HSCT) - most common secondary cause; resembles chronic graft-versus-host disease
- Lung transplantation
Drugs/Chemotherapy:
- Alkylating agents: carmustine, cyclophosphamide
- Amiodarone
- Bleomycin
Autoimmune/Connective Tissue Diseases:
- Rheumatoid arthritis (RA)
- Systemic sclerosis / primary Sjogren's syndrome
- Ankylosing spondylitis, ulcerative colitis
Infections:
- Recurrent pulmonary infections (strong history in most patients)
- Aspergillus, Mycobacterium avium intracellulare
Genetic:
- Familial cases described (sibling pairs), suggesting genetic predisposition
- Mutations in telomere-regulating genes (TERT - telomere reverse transcriptase; TERC - telomerase RNA) identified; associated with more progressive disease
Environmental:
- Aluminosilicate dust, asbestos exposure implicated
- No association with cigarette smoking (a key distinguishing point)
4. Epidemiology
- Age of presentation: highly variable, 8 to 87 years; bimodal distribution - smaller peak in the 3rd decade, larger peak in the 6th decade
- No sex predilection overall (some reports show female predominance especially in young patients)
- Mean time from symptom onset to diagnosis: 2-3 years
- Most patients are non-smokers with a history of recurrent pulmonary infections
5. Clinical Features
Symptoms:
- Progressive dyspnea on exertion (most common)
- Cough (may or may not be present)
- Weight loss (worsens with disease progression)
Physical Examination:
- Often normal in early disease
- Platythorax (flat chest): characteristic finding - reduced anteroposterior diameter of the thorax, visible as deepening of the suprasternal notch. Ratio of AP to transverse diameter is abnormally low. May worsen over time.
- Crackles appear when PPFE extends beyond upper zones, or when coexisting UIP/NSIP/HP is present
- Clubbing: in less than 25% of patients
Complications:
- Spontaneous pneumothorax and pneumomediastinum - can be asymptomatic and recurrent; occur in both idiopathic and secondary forms
6. Laboratory Features
- No diagnostic laboratory tests exist for PPFE
- Elevated KL-6 (Krebs von den Lungen-6) antigen in some patients
- Elevated Surfactant Protein D (SP-D) in some
- Elevated urinary desmosine (a cross-link marker of elastin) found in biopsy-proven PPFE - not yet validated for clinical use
7. Pulmonary Function Tests (PFTs)
- Restrictive ventilatory defect: reduced TLC, decreased FVC, increased FEV1/FVC ratio
- Decreased DLCO
- FVC may decline rapidly, especially in those with telomere gene mutations
- A key distinguishing feature: decrease in FVC out of proportion to DLCO (in early disease, the main defect is pleural fibrosis - extrapulmonary restriction)
- Early stages may show hypercapnia with a normal A-a oxygen gradient (because the early defect is extrapulmonary/pleural restriction, not parenchymal gas exchange impairment)
- Residual volumes may be reduced; mixed ventilatory defects also possible
- Platythorax contributes to ventilation-perfusion mismatch as disease progresses
8. Radiological Features
Chest X-Ray:
- Early disease: may be normal, or show bilateral irregular thickening of apices
- With progression: reticular and nodular opacities in upper lung fields + bilateral hilar opacities
- Lateral view: narrowed AP dimension (platythorax)
- Upward retraction of hila due to upper lobe volume loss
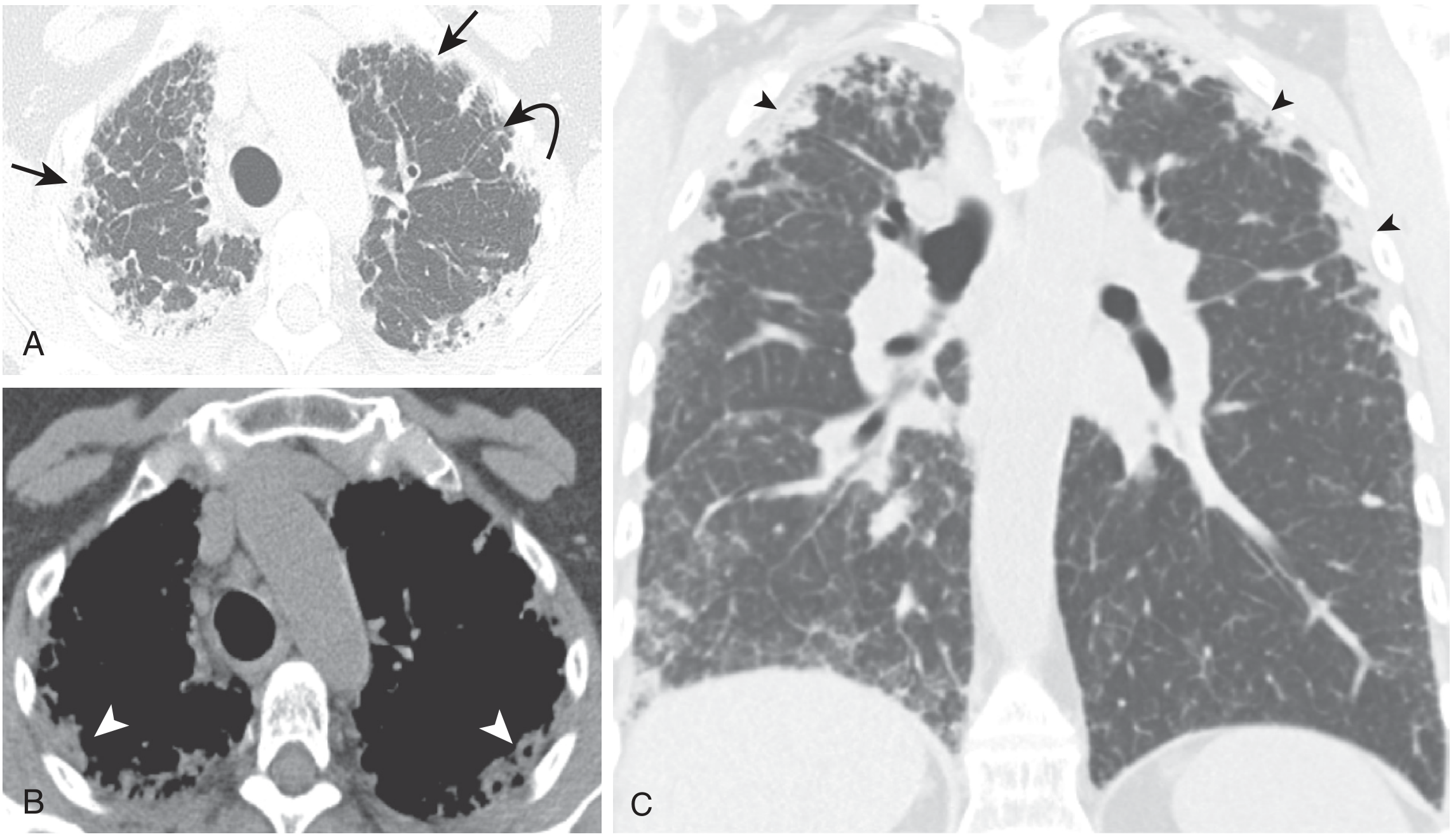
HRCT (Diagnostic Hallmark):
CT findings (Reddy et al. 2012 diagnostic criteria for definitive PPFE):
- Upper lobe pleural thickening with subpleural fibrosis (required)
- Limited lower lobe involvement (required)
- Dense subpleural consolidations with traction bronchiectasis and architectural distortion - bilaterally in upper lobes
- Thickening of the pleura adjacent to subpleural fibrotic changes
- Sharp/clear delineation between areas of fibrosis and normal lung (characteristic)
- Volume loss with upward hilar retraction
- Cysts and bullae may be present
- Coexistent lower lobe changes in UIP-like or NSIP-like pattern are increasingly reported
Key CT images from Murray & Nadel (Figure 90.12):
9. Histopathological Features
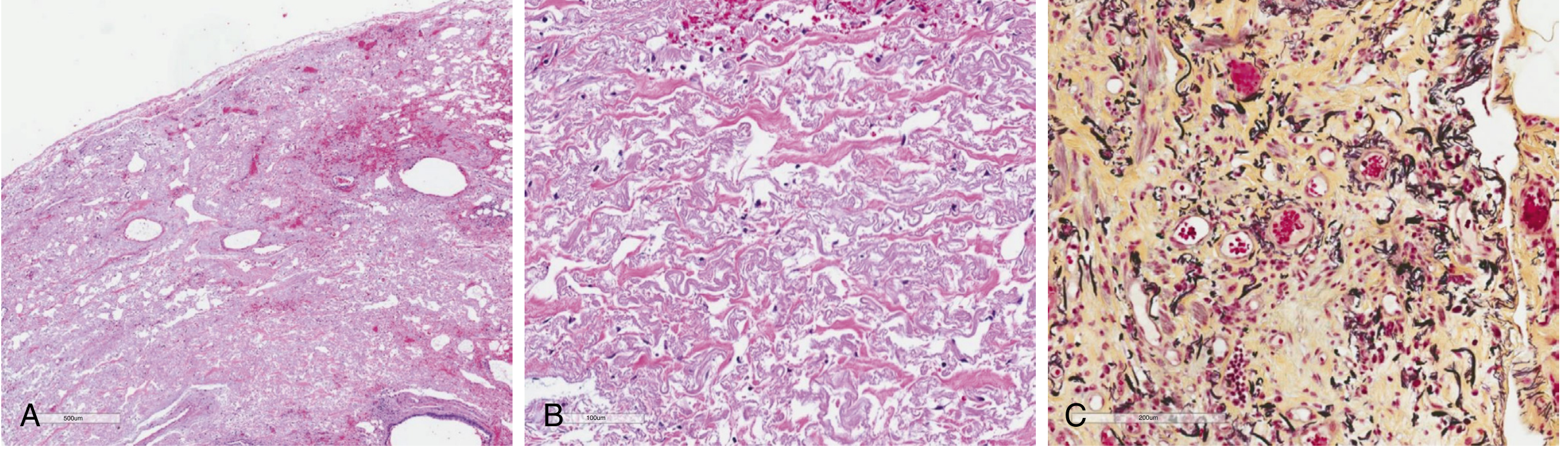
The triad of histopathological findings:
- Visceral pleural fibrosis - dense collagenous fibrosis of the visceral pleura, upper zone predominant (but all zones may be involved histologically even when not apparent on CT)
- Subpleural intra-alveolar fibrosis and elastosis (IAFE) - fibrosis of subpleural lung with septal elastosis
- Elastic fiber deposition - disordered elastic fibers with intervening collagen within alveolar septa
Key histological characteristics:
- Sharp delineation between involved subpleural region and normal lung parenchyma (homogeneous fibroelastosis)
- Rare fibroblastic foci may be present at the leading edge adjacent to normal parenchyma
- Upper lobe-predominant fibrotic and elastotic changes
- IAFE is not exclusive to PPFE (can also be seen in paraquat toxicity, radiation injury, ARDS, apical cap)
Special stains:
- H&E: Pink collagenous fibrous tissue with disordered elastic fibers
- Pentachrome (Movat) stain: mature collagen stains yellow; irregular, fragmented elastic fibers stain black
10. Diagnosis
A multidisciplinary approach is the gold standard, involving clinicians, radiologists, and pathologists with ILD expertise.
Diagnostic approach:
- Clinical history + CT findings are often sufficient for confident diagnosis
- Transbronchial biopsy or transthoracic (CT-guided) biopsy: used when clinical/imaging data are insufficient; non-surgical sampling is preferred
- Surgical lung biopsy (SLB): can be used but is often avoided due to complication risk, especially in secondary PPFE
Differential Diagnosis includes:
- Apical cap (localized fibrotic lesion vs. bilateral PPFE)
- Sarcoidosis (upper lobe predominance but different CT pattern)
- HP (can coexist)
- UIP/IPF (predominantly lower lobe)
- Post-radiation/paraquat changes
Biomarkers: KL-6, SP-D may be elevated but are nonspecific.
11. Treatment
No proven effective treatment exists for PPFE.
- Corticosteroids and immunosuppressive therapy: generally not beneficial - a case series of 12 patients showed prednisone (with or without additional immunosuppression) had no beneficial effect
- Antifibrotic drugs (pirfenidone, nintedanib): currently under investigation; no established evidence yet
- Pleurodesis: may be considered for recurrent pneumothorax
- Lung transplantation: may be required in severe, progressive disease; however, PPFE can recur post-transplant, and secondary PPFE from transplantation itself is recognized
- Supportive care: oxygen therapy, pulmonary rehabilitation
Key exam point: Standardization of diagnostic criteria and prospective research are urgently needed in this disease. - Murray & Nadel
12. Prognosis and Clinical Course
- Progressive disease in most patients
- In a series of 52 patients with idiopathic PPFE diagnosed after multidisciplinary discussion: 5-year mortality was 42%
- In patients with coexisting IIP (especially lower lobe UIP): more rapidly progressive course
- FVC decline is especially marked in patients with telomere gene abnormalities
- Some experience an indolent course; others have rapidly progressive and often fatal disease
- Recurrent pneumothorax is associated with higher morbidity
Summary Table for Quick Revision
| Feature | Key Points |
|---|---|
| Classification | Rare IIP (ATS/ERS 2013) |
| Pathology | Visceral pleural fibrosis + IAFE + elastosis; upper zone predominant |
| CT hallmark | Bilateral upper lobe subpleural consolidation + pleural thickening + sharp demarcation from normal lung |
| Key physical sign | Platythorax (flat chest) |
| PFT pattern | Restrictive; FVC drop out of proportion to DLCO; early hypercapnia with normal A-a gradient |
| Complications | Spontaneous/recurrent pneumothorax |
| Secondary causes | HSCT, lung Tx, alkylating agents, amiodarone, RA, Sjogren's, telomere gene mutations |
| Smoking | NOT associated (important negative) |
| Treatment | No proven therapy; steroids ineffective; lung Tx in severe cases |
| Prognosis | 5-year mortality ~42%; worse with coexisting UIP or telomere mutations |
| Stain | Movat pentachrome - collagen (yellow), elastin (black) |
| Biomarkers | KL-6, SP-D elevated; urinary desmosine (experimental) |
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine (Chapters 90 & 112)
- Fishman's Pulmonary Diseases and Disorders (Chapter 55)
- Goldman-Cecil Medicine (Rare Interstitial Pneumonias)
- Recent reviews: Cottin V et al., Curr Opin Pulm Med 2022 [PMID 35855575]; Yamakawa H et al., J Clin Med 2024 [PMID 38999241]; Ishii H et al., Expert Rev Respir Med 2025 [PMID 40289399]
This is a shared conversation. Sign in to Orris to start your own chat.