Difference between constitutive and inducible enzyme
Reading File
Finding Sources
Searching the Web
constitutive vs inducible enzyme difference biochemistry
Finding Sources
Reading File
Reading File
Here is a clear, comprehensive comparison between constitutive and inducible enzymes:
Constitutive vs. Inducible Enzymes
Definitions
| Feature | Constitutive Enzyme | Inducible Enzyme |
|---|---|---|
| Definition | Produced continuously at a relatively constant level regardless of metabolic state or environmental conditions | Synthesized only under specific conditions, in response to a substrate (inducer) or hormonal/environmental signal |
| Gene expression | Continuously transcribed and translated | Gene is normally repressed; transcription is activated (de-repressed) by an inducer |
| Regulation | Little or no change in amount; regulated mainly by allosteric activity/covalent modification | Regulated primarily at the level of gene transcription (amount of enzyme protein changes) |
| Energy cost | Higher baseline cost - enzyme is always present | More energy-efficient - enzyme is made only when needed |
| Response time | Immediately available | Lag period required (time for new protein synthesis) |
| Examples | Hexokinase (most tissues), COX-1, eNOS, glucocerebrosidase, housekeeping enzymes of glycolysis | Glucokinase (induced by insulin/glucose), COX-2 (induced by inflammation), iNOS (induced by cytokines), PEPCK (induced by glucagon/glucocorticoids) |
Key Characteristics
Constitutive enzymes:
- Carry out essential "housekeeping" metabolic functions that must occur at all times (e.g., glycolysis, lysosomal digestion)
- COX-1 is the classic example - it constitutively produces prostaglandins for gastric cytoprotection, platelet aggregation, and vascular homeostasis, even in platelets that lack nuclei and cannot produce an inducible enzyme on demand (as noted in Firestein & Kelley's Textbook of Rheumatology)
- eNOS (endothelial NOS) is constitutive, producing low, steady levels of NO for physiologic vascular tone regulation (Basic Medical Biochemistry - A Clinical Approach, 6e)
- Defects in constitutive enzymes cause disease even without any environmental trigger (e.g., glucocerebrosidase deficiency in Gaucher disease)
Inducible enzymes:
- Expressed only when the cell encounters a specific signal - substrate, hormone, or inflammatory mediator
- Glucokinase (liver) is induced by insulin in the fed state; its concentration rises with high blood glucose and falls during fasting (Basic Medical Biochemistry - A Clinical Approach, 6e)
- COX-2 is induced by inflammatory cytokines and mediates pain, fever, and inflammation - this is why selective COX-2 inhibitors (e.g., celecoxib) are anti-inflammatory while sparing the gastric mucosa
- iNOS is induced by pro-inflammatory cytokines (e.g., LPS, TNF-α), producing large amounts of NO that drive vasodilation and hypotension in sepsis
- PEPCK and Glucose-6-phosphatase are induced during fasting (by glucagon, glucocorticoids) to upregulate gluconeogenesis
Classic Example: The lac Operon (Microbiology)
The lac operon of E. coli is the textbook model of enzyme induction:
- In the absence of lactose, a repressor protein blocks transcription of beta-galactosidase, permease, and transacetylase
- When lactose is present, an inducer molecule (allolactose) binds the repressor, releasing it from the operator and allowing transcription to proceed
- This is the prototype for substrate-induced enzyme synthesis
Clinical Relevance
| Scenario | Enzyme Type | Significance |
|---|---|---|
| NSAIDs and gastric ulcers | COX-1 (constitutive) inhibited | Loss of gastric protection |
| Selective COX-2 inhibitors | COX-2 (inducible) targeted | Anti-inflammatory without GI damage |
| Sepsis/shock | iNOS (inducible) overactivated | Massive NO production → hypotension |
| Gaucher disease | Glucocerebrosidase (constitutive) deficient | Lipid accumulation in macrophages |
| Antibiotic resistance | Methylase can be constitutive or macrolide-inducible | Determines cross-resistance between clindamycin and macrolides (Goodman & Gilman's) |
In summary: Constitutive enzymes are always "on" for housekeeping roles; inducible enzymes are switched "on" only when their specific signal is present, making them adaptive but with a built-in lag for new protein synthesis.
Acetaminophen
Reading File
Finding Sources
Searching the Web
acetaminophen mechanism of action pharmacology toxicity 2025
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Excellent - I now have everything needed for a comprehensive answer. Here it is:
Acetaminophen (Paracetamol / APAP)
1. Basic Profile
- Class: Non-opioid analgesic and antipyretic (not an NSAID - lacks significant anti-inflammatory activity)
- Other names: Paracetamol (international), Tylenol, Panadol
- Active metabolite of phenacetin (phenacetin was withdrawn due to nephrotoxicity; acetaminophen retains the analgesic effect without causing interstitial nephritis or papillary necrosis)
2. Mechanism of Action
The exact mechanism remains incompletely understood, but several pathways are recognized:
- Central COX inhibition: Acetaminophen inhibits prostaglandin E2 (PGE2) synthesis in the CNS, either via direct COX-2 inhibition or inhibition of membrane-associated prostaglandin synthase. It is a weak COX-1 and COX-2 inhibitor in peripheral tissues - this is why it has no significant anti-inflammatory effect peripherally (Katzung's Basic and Clinical Pharmacology)
- COX-3 variant: It has been proposed to inhibit a COX-1 variant (sometimes called COX-3), especially for thermoregulation (ROSEN's Emergency Medicine)
- Endocannabinoid/serotonergic pathway: The metabolite AM404 (formed in the CNS) activates cannabinoid CB1 receptors and TRPV1 vanilloid receptors, contributing to analgesia. Interactions with the serotonergic system are also described (Morgan & Mikhail's Clinical Anesthesiology)
Key point: Unlike NSAIDs, acetaminophen does NOT inhibit peripheral COX, does not affect platelet function, does not cause gastric ulceration, and has no uricosuric or antiplatelet effects.
3. Pharmacokinetics
| Parameter | Details |
|---|---|
| Administration | Oral, rectal, IV (IV formulation available) |
| Absorption | Rapid; peak plasma concentration within 1 hour, complete by 4 hours |
| Protein binding | Low (poorly bound) |
| Half-life | 2-3 hours (therapeutic doses); may double with liver disease or overdose |
| Excretion | Primarily renal (as conjugates); <5% unchanged |
4. Metabolism - The Critical Concept
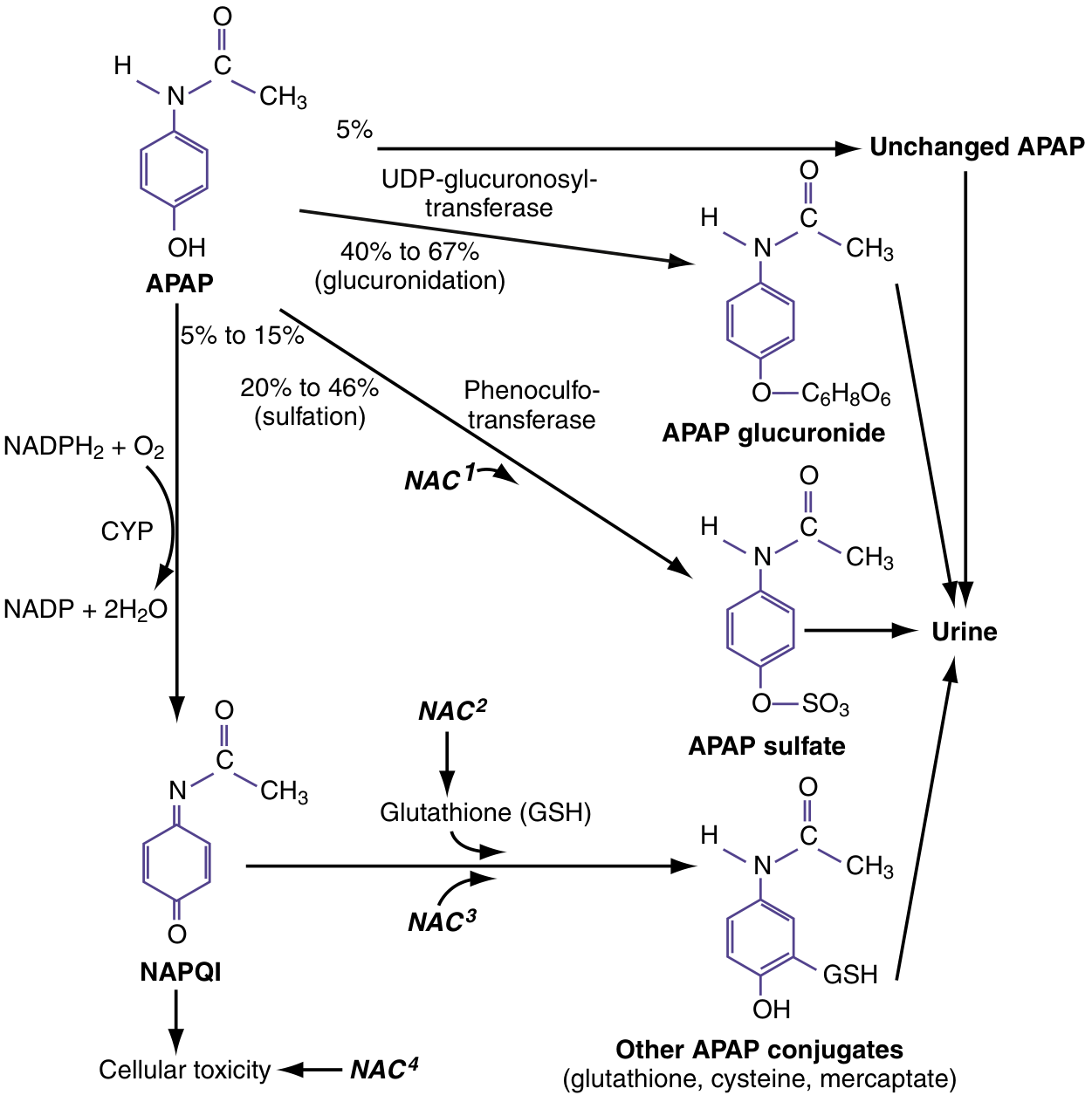
In therapeutic doses, acetaminophen is metabolized via three routes:
- Glucuronidation (40-67%) - by UDP-glucuronosyltransferase → nontoxic APAP glucuronide → excreted in urine
- Sulfation (20-46%) - by phenosulfotransferase → nontoxic APAP sulfate → excreted in urine
- CYP oxidation (5-15%) - primarily by CYP2E1 (also CYP1A2 and CYP3A4) → NAPQI (N-acetyl-p-benzoquinone imine), a highly reactive, cytotoxic electrophile
- In therapeutic doses, NAPQI is rapidly detoxified by conjugation with glutathione (GSH) to form nontoxic mercaptate and cysteine conjugates → excreted in urine
In overdose, glucuronide and sulfate pathways become saturated, and more acetaminophen is shunted to CYP oxidation. NAPQI production overwhelms GSH stores. Unbound NAPQI covalently binds to hepatocyte proteins → initiating cell death cascade.
Source: ROSEN's Emergency Medicine; Katzung's Basic and Clinical Pharmacology
5. Indications
- Mild to moderate pain (headache, myalgia, osteoarthritis, postoperative pain)
- Fever (antipyresis)
- Preferred over aspirin in: patients with hemophilia, peptic ulcer disease, aspirin-sensitive asthma/bronchospasm, children with viral illness (avoids Reye syndrome risk)
- Often combined with opioids for moderate-severe pain (opioid-sparing effect)
- Safe in pregnancy (though recent data suggest possible concerns with prolonged use)
6. Dosing
| Population | Dose | Max Daily |
|---|---|---|
| Adults | 325-1000 mg every 4-6 hours | 4 g/day (3 g/day in elderly or those with liver risk) |
| Children | 10-15 mg/kg every 4-6 hours | 75 mg/kg/day (up to 3750 mg) |
| Liver disease / alcoholism | Use with caution or avoid | Reduce to 2 g/day |
7. Toxicity - Hepatotoxicity (Most Important Adverse Effect)
Toxic Doses
- Minimum hepatotoxic single dose: 7.5-10 g (adults); 150 mg/kg (children)
- Severe hepatotoxicity: typically requires >15-25 g
- Chronic alcoholics: as little as 2-6 g/day can be fatal
Mechanism of Hepatotoxicity
- NAPQI accumulates → depletes GSH → covalently binds to hepatocyte proteins → centrilobular (zone 3) necrosis (because CYP2E1 is concentrated in zone 3)
- With severe toxicity: necrosis extends to submassive/panacinar pattern
Risk Factors for Hepatotoxicity (Sleisenger & Fordtran's)
| Factor | Effect |
|---|---|
| Chronic alcohol use | Induces CYP2E1 ↑ NAPQI; depletes GSH; lowers toxic threshold |
| Fasting / malnutrition | Depletes GSH; lowers toxic threshold |
| Isoniazid, phenytoin, phenobarbital | Induce CYP enzymes → ↑ NAPQI |
| Zidovudine | Competes with glucuronidation → more NAPQI |
| Age | Children more resistant than adults |
| Late presentation | Delay >16 hours worsens outcome |
Clinical Stages of Overdose
| Stage | Timing | Features |
|---|---|---|
| I | 0-24 hrs | Nausea, vomiting, malaise, diaphoresis - often asymptomatic |
| II | 24-72 hrs | RUQ pain, rising ALT/AST, coagulopathy begins |
| III | 72-96 hrs | Peak hepatotoxicity - AST/ALT peak (often 2000-10,000 U/L), jaundice, coagulopathy, encephalopathy |
| IV | 4-14 days | Recovery OR death from fulminant hepatic failure |
Survivors recover completely without fibrosis (complete histologic resolution).
8. Rumack-Matthew Nomogram
Used to determine risk of hepatotoxicity and guide NAC therapy after single acute ingestion. Serum acetaminophen level is measured at 4 hours post-ingestion and plotted against time:
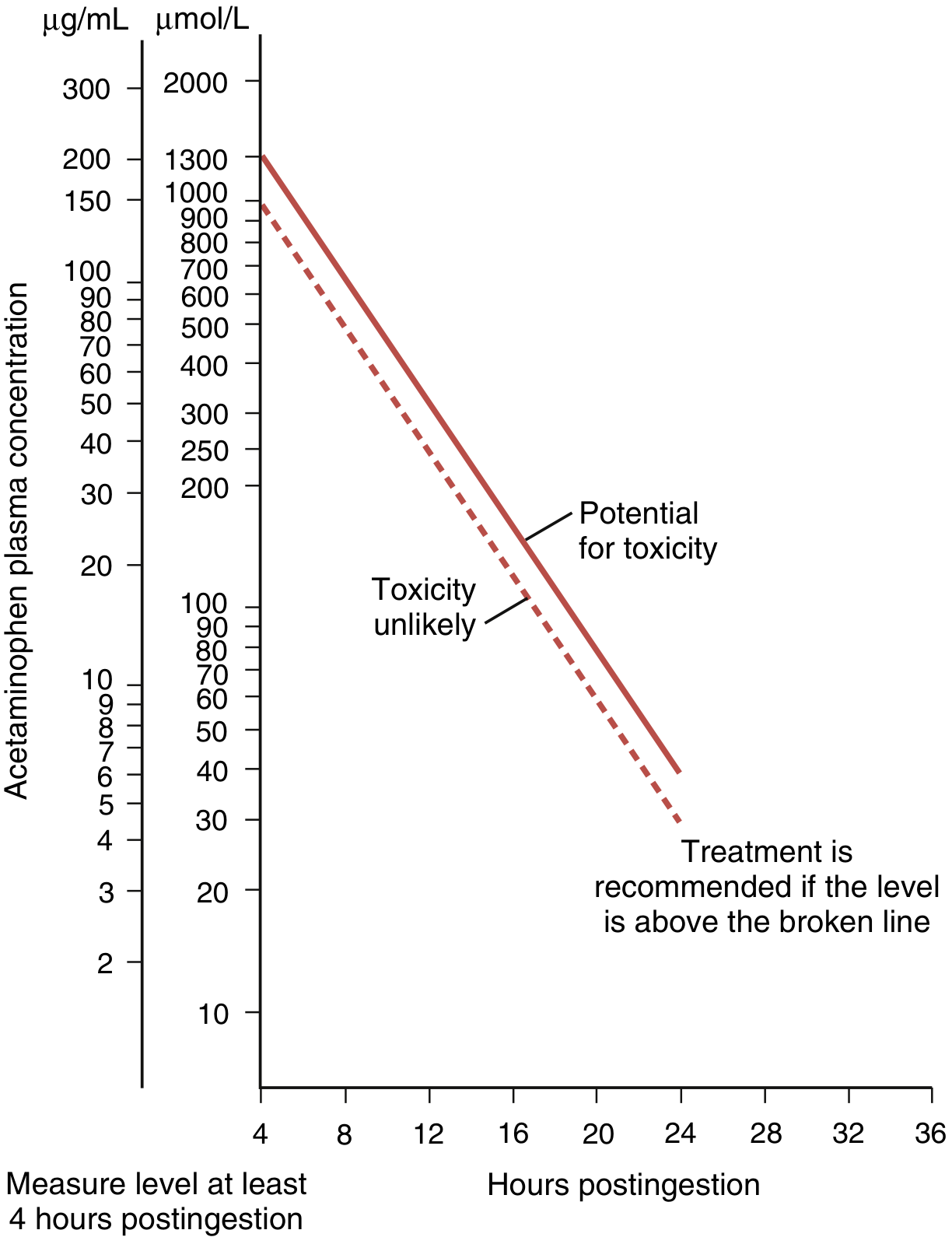
- Level above the treatment line → NAC indicated
- Not applicable for chronic/repeated ingestions
9. Antidote: N-Acetylcysteine (NAC)
NAC works via four mechanisms (see metabolism diagram above):
- NAC¹ - Enhances sulfation (diverts APAP away from toxic pathway)
- NAC² - Serves as glutathione precursor (replenishes GSH stores)
- NAC³ - Acts as a direct glutathione substitute, binding NAPQI directly
- NAC⁴ - Free-radical scavenger; improves hepatic microcirculation and oxygen delivery even after hepatotoxicity is established
NAC Dosing Regimens
Oral (US): Loading dose 140 mg/kg → then 70 mg/kg every 4 hours for 72 hours
IV (Europe/Australia/FDA-approved):
- 150 mg/kg over 15 min in 200 mL D5W
- Then 50 mg/kg over 4 hours
- Then 100 mg/kg over 16 hours (total ~300 mg/kg over 24 hours)
Timing of NAC
| Window | Outcome |
|---|---|
| Within 8 hours | Maximum benefit; near-complete prevention of hepatotoxicity |
| 8-16 hours | Still effective; start immediately without delay |
| 16-36 hours | Hepatotoxicity likely already established, but NAC still reduces mortality (stabilizes vascular reactivity in ALF) |
| >36 hours | Late benefit limited but continue if liver injury present |
Key rule: For maximum benefit, NAC must not be delayed beyond 8 hours after ingestion (ROSEN's Emergency Medicine). If >8 hours have passed, initiate NAC immediately without waiting for labs.
NAC is safe in pregnancy and used at the same doses as non-pregnant patients.
10. Other Adverse Effects
- Renal: Acute tubular necrosis (with or without liver injury); mediated by renal CYP enzymes
- Rare: Hemolytic anemia, methemoglobinemia, anaphylaxis, serious skin reactions (Stevens-Johnson, DRESS) - very rare
- Hepatic enzyme elevation: Mild reversible rise in LFTs even at therapeutic doses
- No platelet effects, no GI ulceration, no uric acid effects
11. Contraindications / Cautions
- Active liver disease or cirrhosis (reduce dose, use with caution)
- Significant alcohol use disorder (lower threshold for toxicity)
- Malnutrition / prolonged fasting (depleted GSH)
- Concurrent use of enzyme-inducing drugs (isoniazid, phenytoin, carbamazepine, rifampicin)
- G6PD deficiency (risk of hemolysis with large doses)
Summary mnemonic for NAPQI toxicity: "Zone 3 necrosis, GSH depletion, CYP2E1, treat with NAC within 8 hours."
Sources: Katzung's Basic and Clinical Pharmacology 16e; ROSEN's Emergency Medicine; Sleisenger & Fordtran's GI and Liver Disease; Fischer's Mastery of Surgery 8e; Morgan & Mikhail's Clinical Anesthesiology 7e
This is a shared conversation. Sign in to Orris to start your own chat.