Diabetes miltus pathophysiology
Reading File
Finding Sources
Finding Sources
Searching PubMed
"diabetes mellitus"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the content needed for a comprehensive answer. Here is the full pathophysiology of Diabetes Mellitus:
Diabetes Mellitus - Pathophysiology
Diabetes mellitus (DM) is a heterogeneous spectrum of metabolic disorders arising from disparate genetic and environmental factors, with a common outcome of impaired glucose homeostasis and hyperglycemia. The pathogenesis involves some combination of insufficient insulin secretion, reduced responsiveness to insulin, increased glucose production, and abnormalities in fat and protein metabolism.
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, p. 2301
1. Normal Glucose Homeostasis (Foundation)
Before discussing pathophysiology, understanding normal physiology is essential.
The Islets of Langerhans are the functional endocrine units of the pancreas:
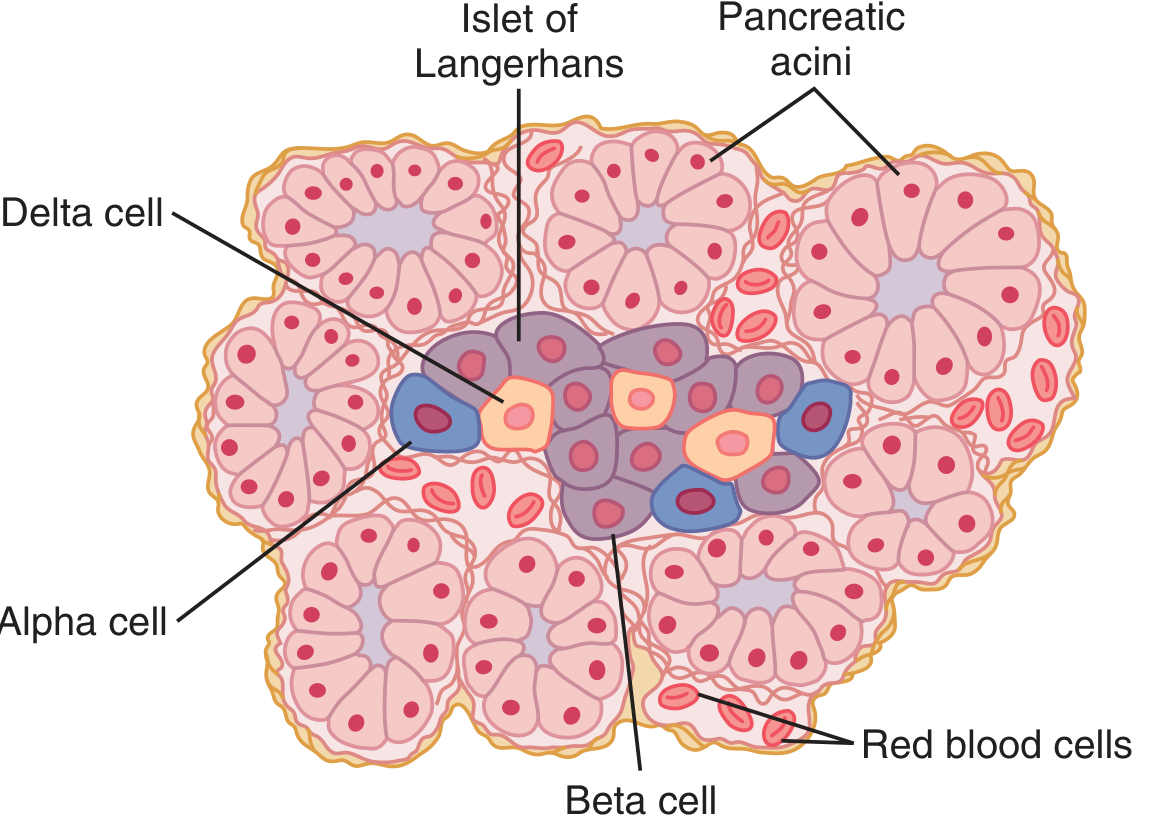
The human pancreas contains 1-2 million islets, each ~0.3 mm in diameter. Three major cell types:
- Beta (β) cells - ~60% of cells; secrete insulin and amylin; located in the islet center
- Alpha (α) cells - ~25%; secrete glucagon
- Delta (δ) cells - ~10%; secrete somatostatin (inhibits both insulin and glucagon)
These cells communicate directly with each other: insulin inhibits glucagon secretion, amylin inhibits insulin secretion, and somatostatin inhibits both.
- Guyton and Hall Textbook of Medical Physiology, p. 2469-2473
Insulin's key metabolic actions:
| Target | Effect |
|---|---|
| Liver | Activates glucokinase, glycogen synthase; inhibits phosphorylase and gluconeogenesis |
| Skeletal muscle | Increases GLUT4-mediated glucose uptake (15-fold), promotes glycogen storage |
| Adipose tissue | Stimulates lipogenesis; inhibits hormone-sensitive lipase (anti-lipolytic) |
| Protein synthesis | Increases amino acid uptake and translation; inhibits catabolism |
2. Classification of Diabetes Mellitus
| Type | Mechanism |
|---|---|
| Type 1 | β-cell destruction → absolute insulin deficiency |
| Type 2 | Insulin resistance + relative insulin secretory defect |
| MODY | Monogenic β-cell disorders (HNF-4α, glucokinase, HNF-1α, etc.) |
| Other | Endocrinopathies, drugs, exocrine pancreas disease |
| Gestational | Insulin resistance during pregnancy |
- Goodman & Gilman's, p. 2510-2554
3. Pathogenesis of Type 1 Diabetes Mellitus
Core mechanism: Autoimmune destruction of β-cells → absolute insulin deficiency
Steps in the process:
- Genetic susceptibility: HLA class II genes (HLA-DR and HLA-DQ) confer 40-50% of genetic risk. Concordance in identical twins is 60-70%.
- Triggering event: An environmental or infectious agent (most likely enterovirus or similar) triggers the autoimmune response in genetically susceptible individuals.
- Cell-mediated β-cell destruction: Autoreactive T-cells infiltrate the islets. Inflammatory mediators released include TNF-α, interferon-γ, and IL-1, all of which cause β-cell death.
- Progressive β-cell loss: Destruction occurs over months to years. Clinical diabetes appears only when the majority of β-cells are destroyed.
Three ADA-recognized stages:
- Stage 1: Autoimmunity (two or more autoantibodies) + normoglycemia
- Stage 2: Autoimmunity + dysglycemia (impaired fasting glucose / impaired glucose tolerance)
- Stage 3: Autoimmunity + hyperglycemia (symptomatic disease)
Autoantibodies found in Type 1 DM:
- Anti-islet cell antibodies
- Anti-insulin antibodies
- Anti-GAD65 (glutamic acid decarboxylase) antibodies
- Anti-IA-2 antibodies
Key note: >75% of individuals with Type 1 DM have no family member with the condition, and the susceptibility genes are present in a significant portion of the nondiabetic population - confirming that genetic susceptibility alone is insufficient.
LADA (Latent Autoimmune Diabetes of Adults): Some adults with an obese phenotype resembling Type 2 DM harbor islet cell autoantibodies - this variant is called Type 1.5 or LADA, with a slowly progressive course.
- Goodman & Gilman's, p. 2568-2575
4. Pathogenesis of Type 2 Diabetes Mellitus
Core mechanism: Insulin resistance + progressive β-cell dysfunction
Type 2 DM is a heterogeneous syndrome. About 80% of patients are overweight or obese, and the condition develops gradually over years, often preceded by a prediabetic stage.
The "Triumvirate" of Type 2 DM pathophysiology:
A. Insulin Resistance
- Primary sites: liver, skeletal muscle, and adipose tissue
- In skeletal muscle: reduced GLUT4 translocation impairs glucose uptake
- In the liver: failure to suppress hepatic glucose output (gluconeogenesis continues despite hyperinsulinemia)
- In adipose tissue: unrestrained lipolysis raises free fatty acids (FFAs) in plasma
- Excess visceral fat is a major driver - adipose-derived inflammatory cytokines (adipokines, TNF-α, IL-6) further impair insulin signaling
B. Compensatory Hyperinsulinemia
- Initially, β-cells compensate by producing more insulin to overcome peripheral resistance
- This maintains near-normal glucose levels for years
- The "first-phase" insulin secretion (the rapid spike within minutes of glucose ingestion) becomes progressively blunted
C. Progressive β-cell failure
- Over time, β-cells are unable to sustain the compensatory hypersecretion
- Mechanisms of β-cell loss include:
- Glucotoxicity: chronic hyperglycemia impairs β-cell gene expression and function
- Lipotoxicity: excess FFAs cause lipid accumulation in β-cells (lipoapoptosis)
- Amyloid deposition: amylin (IAPP) forms deposits in islets, impairing β-cell function
- Oxidative stress and ER stress
- Once β-cells fail, frank diabetes ensues
Role of Glucagon (the "bihormonal" hypothesis):
In Type 2 DM, alpha-cell regulation is also dysregulated. Glucagon levels fail to suppress postprandially and remain inappropriately elevated, further driving hepatic glucose output - compounding hyperglycemia.
Causes of insulin resistance:
| Cause | Example |
|---|---|
| Obesity/visceral fat | Most common |
| Excess glucocorticoids | Cushing syndrome, steroid therapy |
| Excess growth hormone | Acromegaly |
| Polycystic ovary syndrome | ~80% have insulin resistance |
| Lipodystrophy | Ectopic fat in liver |
| Pregnancy | Gestational DM |
| Autoantibodies to insulin receptor | Rare |
| Genetic mutations (PPARγ, melanocortin receptor) | Rare |
- Goodman & Gilman's, p. 2577-2600; Guyton & Hall, p. 2946-3060
5. Metabolic Consequences of Insulin Deficiency / Resistance
Carbohydrate metabolism:
- Reduced glucose uptake by muscle and fat
- Unopposed hepatic gluconeogenesis and glycogenolysis
- Result: Hyperglycemia
- When blood glucose exceeds the renal threshold (~180 mg/dL): glycosuria and osmotic diuresis → polyuria, polydipsia, dehydration
Fat metabolism:
- Loss of insulin's anti-lipolytic effect on adipose tissue → unrestrained lipolysis
- Massive release of free fatty acids (FFAs) → β-oxidation in liver → excess Acetyl-CoA
- Acetyl-CoA exceeds TCA cycle capacity → ketogenesis:
Acetoacetate → β-hydroxybutyrate → Acetone - Result: Diabetic Ketoacidosis (DKA) - mainly in Type 1 (absolute insulin deficiency)
- In Type 2: enough residual insulin to prevent unrestrained ketogenesis, but may develop Hyperosmolar Hyperglycemic State (HHS)
Protein metabolism:
- Insulin deficiency halts protein synthesis and increases catabolism
- Amino acids released from muscle → used for gluconeogenesis
- Result: muscle wasting, weight loss, increased urinary urea
- Guyton & Hall, p. 2697-2699
6. Diagnostic Criteria for DM
| Test | Normal | Pre-diabetes | Diabetes |
|---|---|---|---|
| Fasting plasma glucose | < 5.6 mmol/L (100 mg/dL) | 5.6-6.9 mmol/L | ≥ 7.0 mmol/L (126 mg/dL) |
| 2-hr OGTT (75g) | < 7.8 mmol/L | 7.8-11.0 mmol/L | ≥ 11.1 mmol/L (200 mg/dL) |
| HbA1c | < 5.7% | 5.7-6.4% | ≥ 6.5% |
| Random glucose + symptoms | - | - | ≥ 11.1 mmol/L (200 mg/dL) |
Adapted from ADA/WHO criteria - Goodman & Gilman's, p. 2458-2490
7. Chronic Complications - Mechanisms
All chronic complications stem from prolonged hyperglycemia damaging blood vessels and nerves through several key mechanisms:
| Mechanism | Result |
|---|---|
| Non-enzymatic glycation of proteins (e.g., HbA1c, collagen, myelin) | Structural protein dysfunction, basement membrane thickening |
| Polyol pathway activation (aldose reductase converts glucose to sorbitol) | Intracellular sorbitol accumulation → osmotic damage in nerves, retina, lens |
| Oxidative stress | Endothelial damage, atherosclerosis |
| Protein kinase C activation | Increased vascular permeability, neovascularization |
| Advanced Glycation End-products (AGEs) | Cross-link collagen, activate receptors (RAGE) → inflammation |
Resulting complications:
-
Retinopathy - microaneurysms, neovascularization → leading cause of blindness
-
Nephropathy - glomerular basement membrane thickening, Kimmelstiel-Wilson nodules → end-stage renal disease
-
Neuropathy - peripheral, autonomic (demyelination, axonal loss)
-
Macrovascular disease - accelerated atherosclerosis → MI, stroke, peripheral vascular disease
-
Guyton & Hall, p. 2937-2938; Goodman & Gilman's, p. 2301
Summary Diagram (Conceptual)
Genetic factors + Environmental triggers
↓
TYPE 1: Autoimmune β-cell destruction
→ Absolute insulin deficiency
→ Hyperglycemia + DKA
TYPE 2: Insulin resistance (liver, muscle, fat)
→ Compensatory hyperinsulinemia
→ Progressive β-cell failure
→ Relative insulin deficiency
→ Hyperglycemia + HHS
BOTH: Chronic hyperglycemia
→ Non-enzymatic glycation / Polyol pathway / AGEs / PKC / Oxidative stress
→ Retinopathy / Nephropathy / Neuropathy / Cardiovascular disease
Sources:
- Goodman & Gilman's The Pharmacological Basis of Therapeutics - Chapter 51: Pathophysiology and Diagnosis of Diabetes Mellitus
- Guyton and Hall Textbook of Medical Physiology - Chapter 79: Insulin, Glucagon, and Diabetes Mellitus
This is a shared conversation. Sign in to Orris to start your own chat.