Explain the pathophysiology of mysthenia gravis
Reading File
Finding Sources
Searching PubMed
"myasthenia gravis"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have all the content needed for a thorough, well-sourced answer. Let me compile it.
Pathophysiology of Myasthenia Gravis
Myasthenia gravis (MG) is an autoimmune disorder of the neuromuscular junction (NMJ). Its hallmark is fatigable skeletal muscle weakness that worsens with activity and improves with rest. Understanding the pathophysiology requires first understanding normal NMJ transmission, then examining how antibody-mediated damage disrupts it.
1. Normal Neuromuscular Junction Transmission
The diagram below shows the normal presynaptic NMJ architecture:
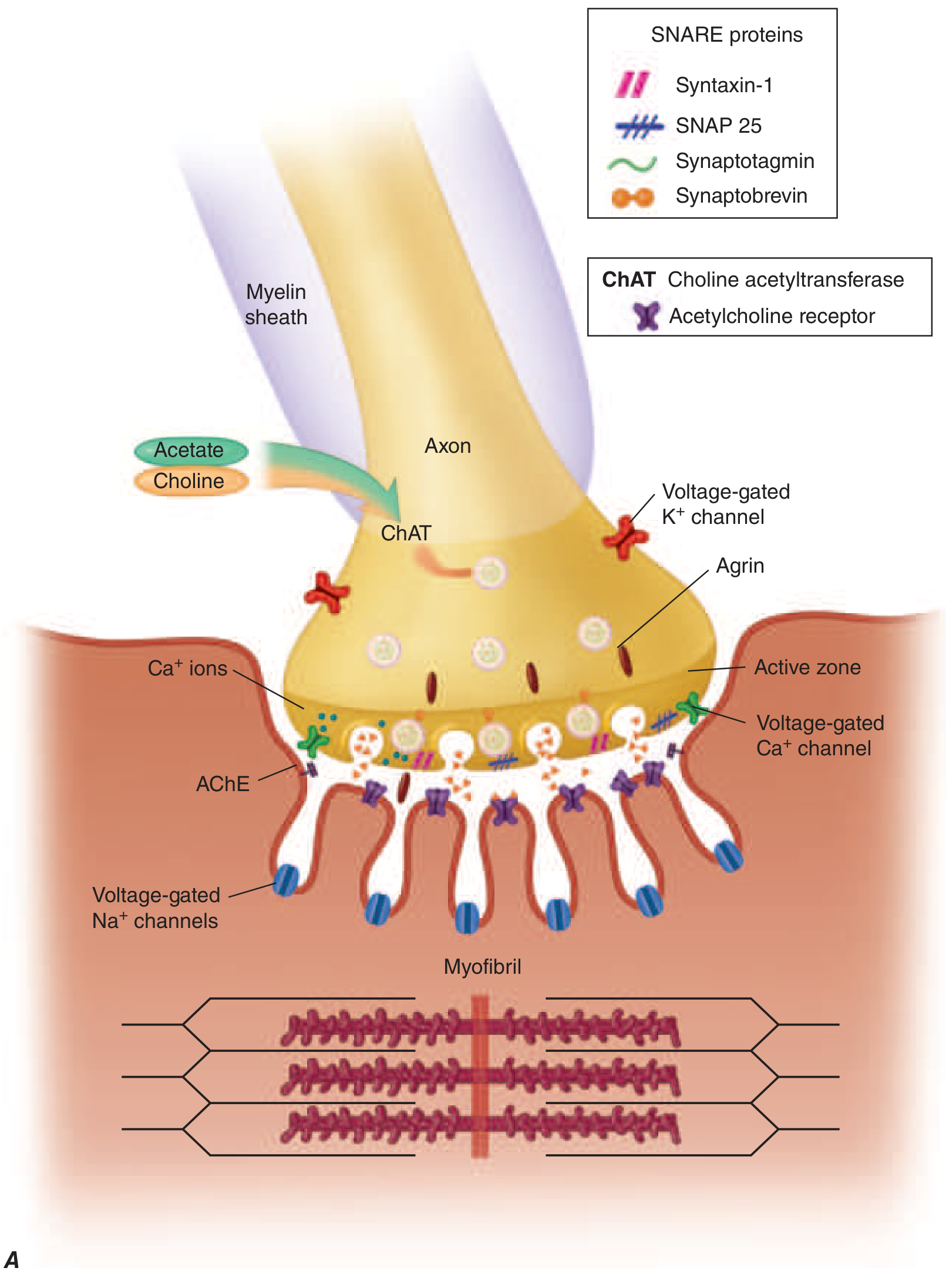
At a normal NMJ, the sequence is:
- An action potential travels down the motor nerve axon to the presynaptic terminal.
- Voltage-gated Ca²⁺ channels open, triggering fusion of 150-200 ACh-containing vesicles with the presynaptic membrane (via SNARE proteins: syntaxin-1, SNAP-25, synaptobrevin, synaptotagmin).
- Released ACh diffuses across the synaptic cleft and binds to nicotinic AChRs densely packed at the crests of postsynaptic folds.
- The AChR is a pentameric ion channel (2α, 1β, 1δ, and 1γ or ε subunits). ACh binds to the α subunits, opening the channel and allowing rapid cation (mainly Na⁺) influx, generating an end-plate potential (EPP).
- If the EPP is large enough, it triggers a muscle action potential and contraction.
- The signal is terminated by acetylcholinesterase (AChE) in the synaptic cleft, which hydrolyzes ACh, and by diffusion away from the receptor.
Stabilizing proteins at the postsynaptic membrane: Agrin (released from the motor nerve terminal) binds LRP4 (lipoprotein receptor-related protein 4). The agrin-LRP4 complex activates MuSK (muscle-specific tyrosine kinase), which recruits rapsyn and DOK7 to cluster and anchor AChRs at the postsynaptic membrane. This scaffolding is critical for efficient synaptic transmission. - Harrison's Principles of Internal Medicine 22E
2. Core Defect in MG
The fundamental defect is a reduction in the number of functional AChRs at the postsynaptic membrane, combined with flattening ("simplification") of the postsynaptic folds. This narrows the safety margin of neuromuscular transmission: ACh is released normally from the nerve terminal, but the resulting EPPs are too small to reliably trigger muscle action potentials. Fewer muscle fibers are activated, producing weakness. - Harrison's Principles of Internal Medicine 22E
Fatigability arises because the amount of ACh released per nerve impulse normally declines with repeated stimulation (presynaptic rundown). In healthy individuals, there is a large safety margin and this doesn't cause failure. In MG, where the AChR reserve is already depleted, this normal rundown means successive impulses activate fewer and fewer muscle fibers, producing progressive weakness with activity. This is also the basis of the decremental response (>10% amplitude drop at 3 Hz) seen on repetitive nerve stimulation (RNS) studies.
3. Autoantibody-Mediated Mechanisms
A. Anti-AChR Antibodies (~85% of generalized MG cases)
These IgG antibodies target the α1 subunit of the nicotinic AChR and damage the postsynaptic membrane via three distinct mechanisms:
| Mechanism | How it works |
|---|---|
| Antigenic modulation (cross-linking and endocytosis) | Anti-AChR antibodies cross-link adjacent receptors, triggering accelerated internalization and lysosomal degradation - reduces receptor density |
| Complement-mediated membrane attack | Antibody binding activates the classical complement pathway, generating the membrane attack complex (MAC) that directly destroys the postsynaptic membrane and its junctional folds |
| Direct blockade | Some antibodies bind directly to the ACh-binding site on the α subunit, competitively blocking ACh from interacting with the receptor |
The net result is a postsynaptic membrane with fewer receptors, simplified architecture, and widened synaptic clefts. - Harrison's Principles of Internal Medicine 22E; Robbins Pathologic Basis of Disease 11E
B. Anti-MuSK Antibodies (~10% of all MG; ~40% of AChR-antibody-negative patients)
MuSK antibodies are predominantly IgG4 (which cannot fix complement), so the mechanism differs from anti-AChR antibodies. They directly interfere with the binding between MuSK and LRP4, disrupting the agrin signaling pathway that is needed for AChR clustering. This leads to dispersal and loss of AChRs from the postsynaptic membrane without direct complement damage. Clinical features differ: anti-MuSK MG tends to produce more prominent facial, bulbar, neck, and respiratory muscle involvement with less ocular involvement. - Harrison's Principles of Internal Medicine 22E; Bradley and Daroff's Neurology in Clinical Practice
C. Anti-LRP4 Antibodies (~1-3% of all MG patients)
LRP4 is the receptor for agrin and co-activates MuSK. Anti-LRP4 antibodies block this interaction, impairing AChR clustering. These patients tend to have milder symptoms. - Bradley and Daroff's Neurology in Clinical Practice
4. Role of the Thymus (Immunopathogenesis)
The thymus plays a central role in triggering and sustaining the autoimmune response:
- ~75% of AChR antibody-positive patients have thymic abnormalities.
- ~65% have thymic hyperplasia: germinal centers with active B cells appear in the thymus, which is not normal. These B cells produce anti-AChR antibodies.
- ~10% have a thymoma (a tumor of thymic epithelial cells). Almost all thymoma-associated MG involves AChR antibodies.
- ~30% of young patients have thymic hyperplasia with B-cell follicles.
Why the thymus? The thymus normally contains "myoid cells" - muscle-like cells that express AChRs on their surface. It is hypothesized that either thymic hyperplasia or thymoma disrupts normal central tolerance, causing autoreactive T cells that recognize AChR epitopes to escape negative selection. These T cells then provide help to B cells in the periphery (and in thymic germinal centers), driving the autoantibody response. The anti-AChR antibodies are T-cell dependent IgG antibodies, confirming helper T cell involvement.
Additionally, a deficiency or dysfunction of regulatory T cells (Tregs) - which normally suppress autoreactive lymphocytes that escape negative selection - contributes to MG pathogenesis. The thymus is the primary source of Tregs, so thymic dysfunction can simultaneously impair suppression of autoreactivity. - Harrison's Principles of Internal Medicine 22E; Robbins Pathologic Basis of Disease 11E
5. Summary: The Chain of Events
Thymic abnormality (hyperplasia/thymoma)
↓
Loss of self-tolerance to AChR antigen
↓
T helper cell activation → B cell stimulation
↓
Production of pathogenic IgG antibodies
(anti-AChR, anti-MuSK, or anti-LRP4)
↓
Attack on postsynaptic NMJ:
• AChR cross-linking → endocytosis/degradation
• Complement MAC → membrane destruction
• Active site blockade
↓
↓ AChRs + simplified/flattened postsynaptic folds
↓
Small EPPs that fail to trigger action potentials
↓
Fatigable muscle weakness
6. Key Differentiating Points
| Feature | MG | Lambert-Eaton Syndrome (for comparison) |
|---|---|---|
| Site | Postsynaptic | Presynaptic |
| Target | AChR, MuSK, LRP4 | Voltage-gated Ca²⁺ channels |
| Weakness pattern | Worsens with activity | Briefly improves with repeated activity |
| Muscles affected | Ocular, bulbar, proximal | Proximal limbs, spares ocular |
| RNS | Decremental | Incremental |
| Association | Thymic disease | Small cell lung cancer (paraneoplastic) |
- Robbins & Kumar Basic Pathology; Bradley and Daroff's Neurology in Clinical Practice
Recent Literature Note
Recent systematic reviews (2025-2026) on MG have focused on clinical overlap syndromes (PMID: 40209696), exercise in MG (PMID: 41781681), and MG in pregnancy (PMID: 41787190). No recent systematic reviews have contradicted the established AChR/MuSK/complement-mediated pathophysiology described above - the framework remains consistent with current understanding.
This is a shared conversation. Sign in to Orris to start your own chat.