note on lymphadenitis briefly
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Here is a concise note on lymphadenitis:
Lymphadenitis
Definition
Inflammation of lymph nodes in response to infection or other inflammatory stimuli. Lymph nodes act as defensive barriers, and any immune response against foreign antigens can cause nodal enlargement (lymphadenopathy).
Classification
1. Acute Nonspecific Lymphadenitis
-
Causes: Pyogenic bacteria, systemic viral infections (especially in children), bacteremia
-
Common sites:
- Cervical — drainage from dental/tonsillar infections
- Axillary/Inguinal — infections of the extremities
- Mesenteric — acute appendicitis, gut inflammation, viral GI infections
-
Clinical features: Enlarged, tender nodes; fever, anorexia, malaise; overlying skin may be red; nodes become fluctuant if abscess forms
-
Morphology:
- Nodes are swollen, gray-red, engorged
- Large reactive germinal centers with numerous mitotic figures
- Neutrophilic infiltrate around follicles and within sinuses
- Severe infection → follicular center necrosis → abscess → draining sinuses
-
Treatment: Directed at the primary focus (e.g., tonsillitis, dental abscess)
2. Chronic Nonspecific Lymphadenitis
Assumes one of three histological patterns:
| Pattern | Mechanism | Key Causes |
|---|---|---|
| Follicular hyperplasia | B-cell activation in germinal centers | Rheumatoid arthritis, toxoplasmosis, early HIV |
| Paracortical hyperplasia | T-cell zone reaction; immunoblasts efface B-cell follicles | EBV, vaccinations, drug reactions (phenytoin) |
| Sinus histiocytosis | Distension of sinusoids; hypertrophy of lining endothelium | Carcinomas draining to regional nodes |
- Chronic cervical lymphadenopathy causes include TB, malignancy (squamous cell carcinoma in older adults), lymphoma, HIV
3. TB Lymphadenitis (Scrofula)
- Most common form of extrapulmonary TB
- Preferentially involves posterior cervical and supraclavicular nodes (painless)
- Occurs often without clinical pulmonary disease
- More common in children and adolescents; slight female predominance
- Disease progression → suppuration → sinus tract formation
- In the USA, scrofula is more often caused by non-tuberculous mycobacteria than M. tuberculosis
4. Filarial Lymphadenitis
- Acute episodes due to dying adult worms (Wuchereria bancrofti, Brugia malayi)
- Painful inguinal node enlargement with erythema, fever, chills; may recur
- Can be complicated by epididymitis/orchitis
- Repeated episodes → lymphatic dilation → chronic lymphedema (elephantiasis)
Key Distinguishing Point: Follicular Hyperplasia vs. Follicular Lymphoma
Reactive follicular hyperplasia is distinguished from follicular lymphoma by:
- Preservation of lymph node architecture
- Variation in size/shape of germinal centers
- Mixed lymphocyte population (varying shapes/sizes)
- Prominent mitotic activity and macrophage phagocytosis (tingible body macrophages)
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Bailey and Love's Surgery 28th Ed.; Goldman-Cecil Medicine
write a note on wilson`s disease
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Wilson's Disease
Also known as: Hepatolenticular degeneration
Definition & Genetics
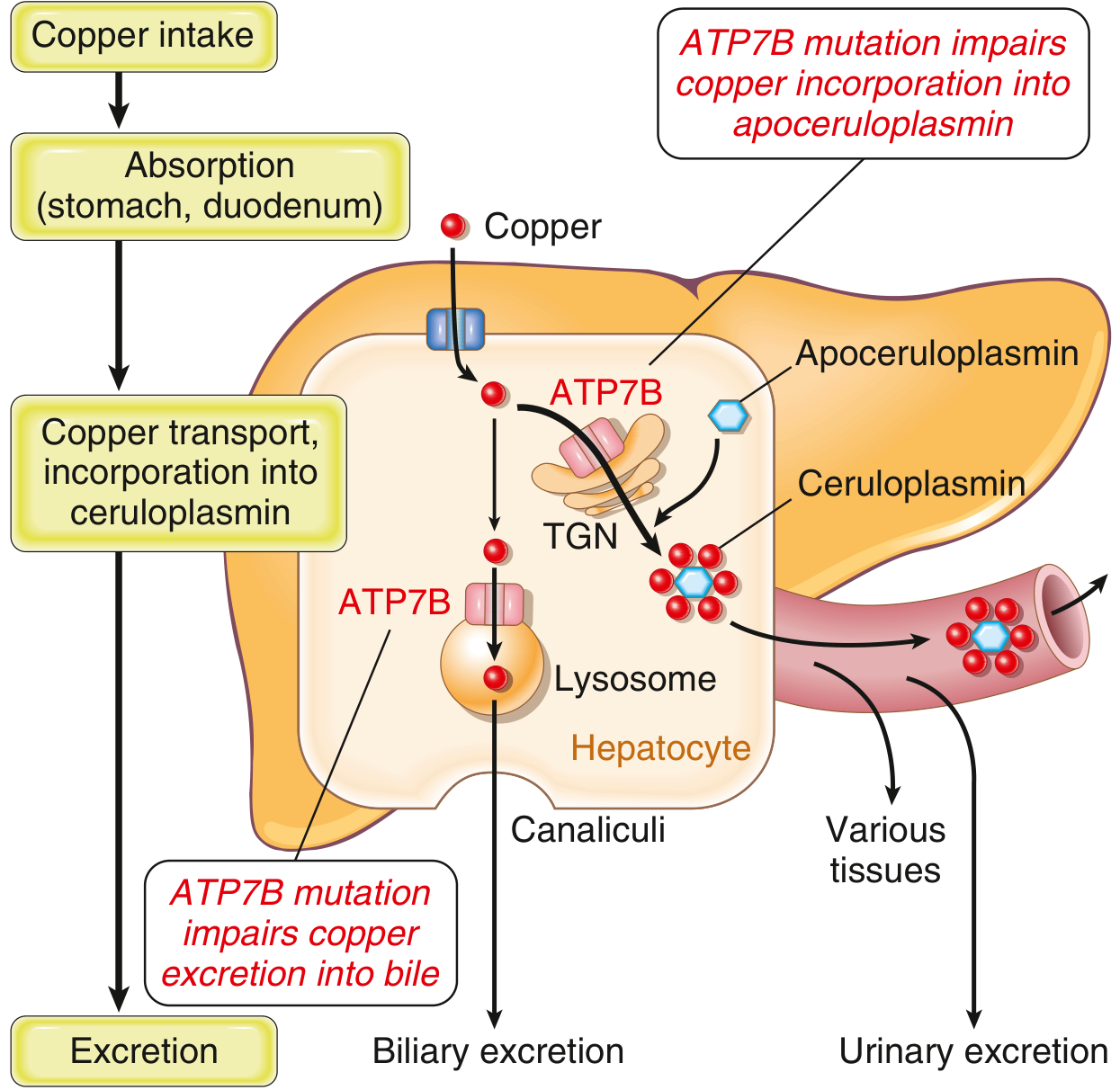
Wilson's disease is an autosomal recessive disorder caused by loss-of-function mutations in the ATP7B gene (chromosome 13q). The ATP7B protein is a copper-transporting P-type ATPase located in the trans-Golgi network and lysosomes of hepatocytes. Mutations impair:
- Copper excretion into bile (major route of copper elimination)
- Copper incorporation into apoceruloplasmin → low serum ceruloplasmin
Result: toxic copper accumulation in liver, brain, cornea, kidneys, bones, and joints.
Epidemiology
- Prevalence ~1 in 30,000
- Symptoms typically appear between 6 and 40 years of age
- Over 300 pathogenic mutations identified; most common: His1069Glu (~40% of cases in certain ethnic groups)
Pathogenesis
Normally, 40–60% of ingested copper (2–5 mg/day) is absorbed in the duodenum and transported to the liver bound to albumin. In hepatocytes, ATP7B:
- Transfers copper to apoceruloplasmin (in trans-Golgi) → secreted as ceruloplasmin into blood
- Transports excess copper via lysosomes into bile canaliculi
In Wilson's disease, both pathways fail → copper accumulates in hepatocyte cytoplasm → ↑ROS → hepatocyte injury → copper spills into blood → deposits in other organs.
Clinical Presentation
About 1/3 present with each of: hepatic disease, neurological features, psychiatric symptoms.
Hepatic (most common initial presentation in children)
| Stage | Features |
|---|---|
| Early | Steatohepatitis, elevated transaminases |
| Intermediate | Chronic active hepatitis, fibrosis |
| Advanced | Cirrhosis, portal hypertension |
| Acute/fulminant | Acute liver failure with Coombs-negative hemolytic anemia; typically in 2nd decade |
Clue for fulminant WD: serum ALP/total bilirubin < 4 AND AST/ALT > 2.2
Neurological (extrapyramidal predominant)
- Tremor (may have wing-beating character), chorea, dystonia
- Dysarthria, dysphagia, ataxia, gait disturbance
- Fixed "sardonic" smile
- Seizures (minority)
- MRI shows abnormal T2 signal in putamen, midbrain, pons, thalamus, cerebellum
Neuropsychiatric
- Present in ≥50% early in the course; ~70% develop symptoms long-term
- Personality/mood changes (most common), depression (~30%), bipolar spectrum (~20%)
- Psychosis; increased sensitivity to neuroleptics
- Suicidal ideation (5–15%)
Ophthalmologic
- Kayser-Fleischer (KF) rings — green-to-brown copper deposits in Descemet membrane at corneal limbus; best seen on slit-lamp
- Present in 98% of neurological WD, 80% of all cases
- Sunflower cataracts — copper in lens
Other organs
- Renal tubular dysfunction (Fanconi syndrome), renal stones
- Hemolytic anemia
- Osteoporosis, arthropathy
- Hypoparathyroidism
Morphology
Liver
| Stage | Histology |
|---|---|
| Early | Glycogen-filled nuclei, microvesicular + macrovesicular steatosis |
| Chronic | Portal mononuclear infiltrates, interface hepatitis, Mallory bodies, periportal fibrosis |
| Fulminant | Coagulative necrosis, microvesicular fat, Kupffer cell pigment, established cirrhosis |
| Late | Mixed micro- and macronodular cirrhosis |
- Electron microscopy: pleomorphic, enlarged mitochondria with widened intracristal spaces (early/characteristic finding)
- Histochemical copper stains unreliable; quantitative liver copper > 250 μg/g dry weight is diagnostic (but only ~80% sensitive)
Brain
- Toxic injury predominantly to basal ganglia (putamen most affected)
Diagnosis
| Test | Finding | Notes |
|---|---|---|
| Serum ceruloplasmin | ↓ (<20 mg/dL) | Hallmark; also low in malnutrition, nephrotic syndrome |
| 24-h urine copper | ↑ (>100 μg/day) | Most specific test |
| Liver copper (biopsy) | >250 μg/g dry wt | Most sensitive test |
| KF rings (slit-lamp) | Present | 98% in neurological WD |
| Serum copper | Variable | Not diagnostically useful |
| MRI brain | T2 hyperintensity basal ganglia | Neurological WD |
| Genetic testing | ATP7B mutation | Useful for family screening; >300 mutations makes it complex |
A scoring system (Leipzig score) assists diagnosis when results are borderline.
Treatment
Wilson's disease is uniformly fatal without therapy. Goals: reduce copper burden + prevent further retention.
Medical
| Indication | Preferred Treatment |
|---|---|
| Initial hepatic disease | Trientine (preferred) or D-penicillamine + zinc |
| Initial neurological | Tetrathiomolybdate (clinical trial), trientine ± zinc |
| Presymptomatic | Zinc or trientine |
| Maintenance | Zinc or trientine |
| Pregnancy | Zinc (safest) |
| Acute liver failure | Plasmapheresis → liver transplantation |
Chelating agents:
- Trientine (triethylenetetramine): now preferred at most centers; better tolerated; dose 10–20 mg/kg/day (1–2 g/day) in 2–3 divided doses before meals
- D-Penicillamine: 1–2 g/day; add pyridoxine 25 mg/day (antipyridoxine effect); 20–50% of neurological patients worsen acutely on initiation
- Start at ¼–½ target dose and titrate over several weeks
Zinc: inhibits intestinal copper absorption by inducing metallothionein in enterocytes; preferred for maintenance and pregnancy
Monitoring: CBC, urinalysis, LFTs weekly for first month; 24-h urinary copper every 6–12 months; target <500 μg/day after several years of chelation.
Surgical
- Liver transplantation — curative; indicated for fulminant liver failure or advanced cirrhosis unresponsive to medical therapy; corrects the metabolic defect
Prognosis
- Early diagnosis and lifelong treatment dramatically alter the course
- Neurological and psychiatric symptoms improve with treatment (but ~70% still develop psychiatric features long-term)
- MRI changes may partially reverse with effective therapy
Sources: Robbins & Kumar Basic Pathology; Yamada's Textbook of Gastroenterology 7th ed.; Bradley and Daroff's Neurology in Clinical Practice; Sleisenger & Fordtran's GI and Liver Disease
This is a shared conversation. Sign in to Orris to start your own chat.