Myocardial infarction
Reading File
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "management"[MeSH Terms]
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "treatment"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full, detailed answer.
Myocardial Infarction (MI)
Definition
MI ("heart attack") is necrosis of cardiac muscle resulting from prolonged ischemia. The 2018 joint ESC/ACC/AHA task force defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia." - Robbins Basic Pathology, p. 357
Approximately 800,000 Americans experience MI annually - nearly one every 40 seconds - causing ~400,000 deaths/year. About 10% occur before age 40 and 45% before age 65.
Pathogenesis
Typical Mechanism (90% of cases)
The classic sequence of events in plaque disruption-mediated MI:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood
- Platelets adhere, aggregate, and activate, releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm
- Coagulation is activated by exposed tissue factor, building the thrombus
- Within minutes, the enlarging thrombus can completely occlude the coronary artery lumen
When angiography is performed within 4 hours of MI onset, coronary thrombosis is demonstrated in nearly 90% of cases. At 12-24 hours (without intervention), only 60% show thrombosis - some occlusions clear spontaneously via thrombus lysis or spasm relaxation. This is the therapeutic rationale for early thrombolysis/angioplasty.
- Robbins & Cotran Pathologic Basis of Disease, p. 511
Atypical Causes (~10%)
- Vasospasm (with or without atherosclerosis) - cocaine, ephedrine
- Embolism from left atrial mural thrombus (in AF), infective endocarditis vegetations, prosthetic material, or paradoxical embolism via patent foramen ovale
- Severe fixed coronary stenosis with a period of increased demand (tachycardia, hypertension) - causes subendocardial infarction
- Small vessel disease: vasculitis, amyloid deposition, sickle cell disease
Coronary Artery Territories and Infarct Sites
| Artery | Frequency | Territory Infarcted |
|---|---|---|
| LAD | 40-50% | Anterior LV wall near apex; anterior ventricular septum; apex circumferentially |
| RCA | 30-40% | Inferior/posterior LV wall; posterior septum; inferior/posterior RV free wall (in some) |
| LCx | 15-20% | Lateral LV wall (except apex) |
- Of RCA occlusions, 15-30% extend into the right ventricular wall. Isolated RV infarction occurs in only 1-3% of cases.
- Robbins & Cotran, p. 512
Morphologic Evolution (Timeline)
The gross and microscopic changes evolve in a predictable sequence:
| Time Frame | Gross Features | Light Microscopy | Key Points |
|---|---|---|---|
| 0-30 min | None | None | Reversible injury; EM shows myofibril relaxation, glycogen loss, mitochondrial swelling |
| 30 min - 4 hr | None | Usually none; wavy fibers at border | Irreversible: sarcolemmal disruption, mitochondrial amorphous densities |
| 4-12 hr | Occasional dark mottling | Coagulative necrosis begins; edema; hemorrhage | - |
| 12-24 hr | Dark mottling | Coagulative necrosis ongoing; nuclear pyknosis; hypereosinophilic myocytes; contraction band necrosis; early neutrophil infiltrate | Gross ID possible as reddish-blue area from congestion |
| 3-7 days | Soft, yellow-tan; hyperemic rim | Neutrophil infiltration peaks, then disappears; macrophage infiltration begins | Rimmed by highly vascularized granulation tissue |
| 1-3 weeks | Yellow-tan, soft, depressed | Granulation tissue replaces necrotic area | Progressive fibrosis |
| >6 weeks | Gray-white scar | Dense fibrous scar | Stable; no regeneration of cardiomyocytes |
Staining trick: Triphenyl tetrazolium chloride (TTC) - viable myocardium stains brick-red (lactate dehydrogenase intact); infarcted zone stays pale/unstained (LDH leaks out). Useful for infarcts >3 hours old.
- Robbins Basic Pathology, p. 357; Robbins & Cotran, p. 512
ECG Changes - Electrophysiology
Three major membrane abnormalities cause ECG changes in acute MI (Ganong's Physiology):
| Defect in Infarcted Cells | Current Flow | ECG Change (leads over infarct) |
|---|---|---|
| Rapid repolarization (accelerated K+ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (loss of intracellular K+) | Into infarct | TQ depression (recorded as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
-
Hallmark of acute MI: ST segment elevation in leads overlying the infarcted area; ST depression in leads on the opposite side
-
After days to weeks: ST changes subside; dead muscle becomes electrically silent
-
Q waves may appear (reflects electrically silent dead tissue during systole)
-
Non-Q-wave infarcts tend to be less severe but have a higher rate of subsequent reinfarction
-
Ganong's Review of Medical Physiology, 26th ed., p. 534
Clinical Features
- Chest pain: severe, crushing/pressure-like, substernal, may radiate to left arm, jaw, or back; typically >20 minutes and not relieved by nitrates
- Diaphoresis, nausea, vomiting
- Dyspnea (from LV dysfunction)
- Sense of impending doom
- Silent MI: common in diabetics and elderly (no chest pain)
Biomarkers:
- Troponin I/T: gold standard; rises within 3-4 hours, peaks at 24-48 hours, elevated for up to 10-14 days
- CK-MB: rises within 4-6 hours, normalizes within 48-72 hours - useful for detecting reinfarction
- Myoglobin: earliest to rise (1-2 hours) but non-specific
Types of MI
- STEMI (ST-elevation MI): full-thickness (transmural) occlusion - requires emergent reperfusion
- NSTEMI (Non-ST-elevation MI): partial occlusion or spontaneous reperfusion - managed with early invasive strategy
- Type 1 MI: plaque rupture/erosion with thrombus
- Type 2 MI: supply-demand mismatch (e.g., severe anemia, tachyarrhythmia, coronary spasm) without primary plaque rupture
Complications
Nearly three-quarters of patients experience one or more complications after acute MI. Overall in-hospital death rate is <7% (STEMI ~9%, NSTEMI ~6%), but out-of-hospital STEMI mortality is ~33% - most die from arrhythmia within 1 hour.
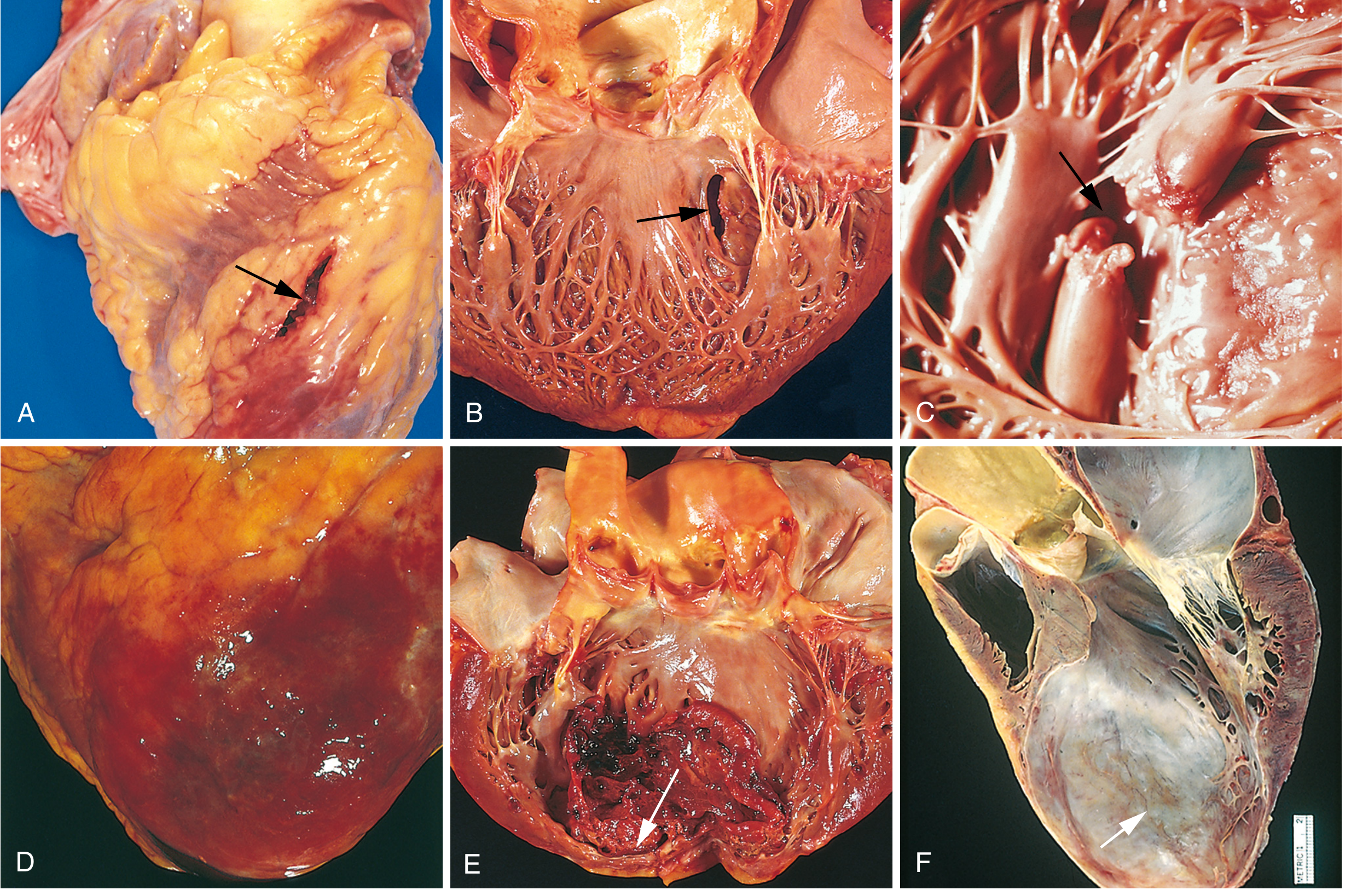
Fig. 12.17 - Robbins & Cotran: Complications of MI
| Complication | Details |
|---|---|
| Contractile dysfunction | LV failure proportional to volume of damage; cardiogenic shock in ~10% of transmural MIs (requires ≥40% LV damage) |
| Papillary muscle dysfunction | Postinfarct mitral regurgitation; papillary muscle rupture is rare but catastrophic |
| RV infarction | With RCA occlusion; leads to right heart failure and systemic hypotension |
| Free wall rupture | 1-3% of MIs; usually fatal hemopericardium and cardiac tamponade (days 3-7, when necrosis is maximal) |
| Ventricular septal rupture | Creates VSD with left-to-right shunt |
| Papillary muscle rupture | Severe acute mitral regurgitation; acute pulmonary edema |
| Mural thrombus | Forms over akinetic endocardium; risk of systemic embolism |
| Ventricular aneurysm | Late complication; paradoxical bulging during systole; predisposes to arrhythmias, mural thrombus, and HF |
| Arrhythmias | Most common early cause of death; VF accounts for most out-of-hospital deaths; reentry circuits from disrupted gap junctions |
| Fibrinous pericarditis | Dressler syndrome (autoimmune) - occurs weeks post-MI |
| Heart block | AV node dysfunction; ranges from 1st degree to complete block |
- Robbins & Cotran Pathologic Basis of Disease, pp. 513-518
Treatment (Acute Management)
STEMI - Reperfusion is the Priority ("Time is Muscle")
- Primary PCI (percutaneous coronary intervention): preferred if available within 90 minutes of first medical contact - balloon inflation within 120 minutes of symptom onset
- Thrombolytics (e.g., alteplase, tenecteplase): if PCI not available within 120 minutes; must be given within 12 hours of symptom onset; contraindicated in prior stroke, active bleeding, recent surgery
Pharmacological Treatment (Both STEMI and NSTEMI)
| Drug Class | Agent | Purpose |
|---|---|---|
| Dual antiplatelet | Aspirin + P2Y12 inhibitor (ticagrelor, clopidogrel, prasugrel) | Prevent further thrombus; ticagrelor/prasugrel preferred over clopidogrel |
| Anticoagulation | Heparin (UFH or LMWH), or bivalirudin | Prevent thrombus extension |
| Beta-blocker | Metoprolol, carvedilol | Reduce heart rate/demand; reduce arrhythmia risk; reduce infarct size |
| ACE inhibitor/ARB | Ramipril, lisinopril | Reduce LV remodeling; mortality benefit especially with reduced EF |
| Statin | Atorvastatin 80 mg | Plaque stabilization; secondary prevention |
| Nitrates | GTN/nitroglycerin | Symptom relief; reduce preload; NOT for hypotension or RV infarction |
| Morphine | IV morphine | Pain relief (use cautiously - may delay P2Y12 absorption) |
| Oxygen | Only if SpO2 <90% | Avoid routine oxygen - may increase infarct size in normoxic patients |
NSTEMI - Antithrombotic and Risk Stratification
- High-risk NSTEMI: early invasive strategy (coronary angiography within 24 hours)
- Low-risk: conservative/ischemia-guided strategy
Secondary Prevention
- Lifelong aspirin + statin
- Beta-blocker for at least 1 year (longer if reduced EF)
- ACE inhibitor/ARB indefinitely if EF <40% or HF
- Aldosterone antagonist (eplerenone) if EF <40% + HF or diabetes
- Cardiac rehabilitation
- Lifestyle modification: smoking cessation, diet, exercise, weight loss
Key Pathophysiology Summary
Plaque rupture/erosion
↓
Platelet adhesion → aggregation → TXA2, ADP, serotonin release
↓
Tissue factor exposure → coagulation cascade
↓
Coronary occlusion → ischemia → ATP depletion
↓
< 20 min: reversible injury
> 20 min: irreversible necrosis
↓
Coagulative necrosis → inflammatory infiltration → scar formation
Sources: Robbins & Cotran Pathologic Basis of Disease (10th ed.); Robbins Basic Pathology (11th ed.); Ganong's Review of Medical Physiology (26th ed.); Braunwald's Heart Disease (12th ed.)
This is a shared conversation. Sign in to Orris to start your own chat.