Step wise pathophysiology of frost bite
frostbite pathophysiology
Stepwise Pathophysiology of Frostbite
Stage 1 - Pre-Freeze: The Hunting Response (Cooling Phase)
- Initial sympathetic vasoconstriction shunts blood away from the periphery to conserve core temperature
- Tissue ischemia triggers reflex vasodilation (a brief "hunting" attempt to rewarm)
- As cooling continues, the hunting response fails and vasoconstriction becomes sustained and permanent
- The extremity shifts toward ambient temperature
- Cold also directly increases blood viscosity and promotes vasospasm
The head has no vasoconstrictor response except the nose and ears, which is why frostbite on the trunk is unusual unless direct contact with refrigerant occurs.
Stage 2 - Ice Crystal Formation (The Freeze)
2a. Extracellular Ice Crystal Formation (Primary Mechanism)
- Ice forms first in the extracellular space (lower solute concentration freezes first)
- Extracellular ice raises the osmotic concentration of the extracellular fluid
- This creates an osmotic gradient that draws water out of cells (cellular dehydration)
- Resulting intracellular hypertonicity causes protein denaturation, enzyme dysfunction, and membrane lipid disorganization
- Cell membranes undergo lysis
2b. Intracellular Ice Crystal Formation (Rapid Freeze)
- Occurs only with very rapid freezing (>10°C/min, or 18°F/min)
- Intracellular ice crystals mechanically rupture organelles and membranes
- This is the most immediately lethal mechanism for the cell
2c. Intravascular Ice Formation
- Ice crystals form within vessel lumens
- Causes erythrocyte sludging and direct vessel occlusion
- Microcirculatory flow ceases
- Electrolyte imbalances (intracellular sodium, calcium overload)
- Failure of Na-K-ATPase pumps
- Cessation of aerobic metabolism
- Progressive cellular metabolic derangements
Stage 3 - Thawing and Reperfusion Injury (The Most Damaging Phase)
3a. Physical Thawing
- Ice crystals melt; dehydrated cells begin to swell
- Vascular wall integrity is compromised (endothelial damage from ice crystals)
- Loss of vascular tone causes sudden vasodilation and pooling
3b. Ischemia-Reperfusion Injury
- Return of blood flow to ischemic tissue generates reactive oxygen species (ROS)
- Oxidative stress causes direct cell membrane damage
- Endothelial injury increases vascular permeability - massive edema ensues
3c. Inflammatory Mediator Release (Peaks Here)
| Mediator | Effect |
|---|---|
| Thromboxane A2 (TXA2) | Potent vasoconstriction + platelet aggregation |
| Prostaglandin F2α (PGF2α) | Vasoconstriction + platelet adhesiveness |
| Bradykinin | Increases vascular permeability |
| Histamine | Vasodilation + permeability |
| Leukotrienes | Leukocyte adhesion, further inflammation |
- Blister fluid contains extremely high concentrations of TXA2 and PGF2α
- These mediators cause further vasoconstriction and extend the injury zone beyond the original freeze
- Platelets and leukocytes aggregate in microvessels, causing progressive thrombosis
- This creates a vicious cycle: ischemia → mediator release → more ischemia
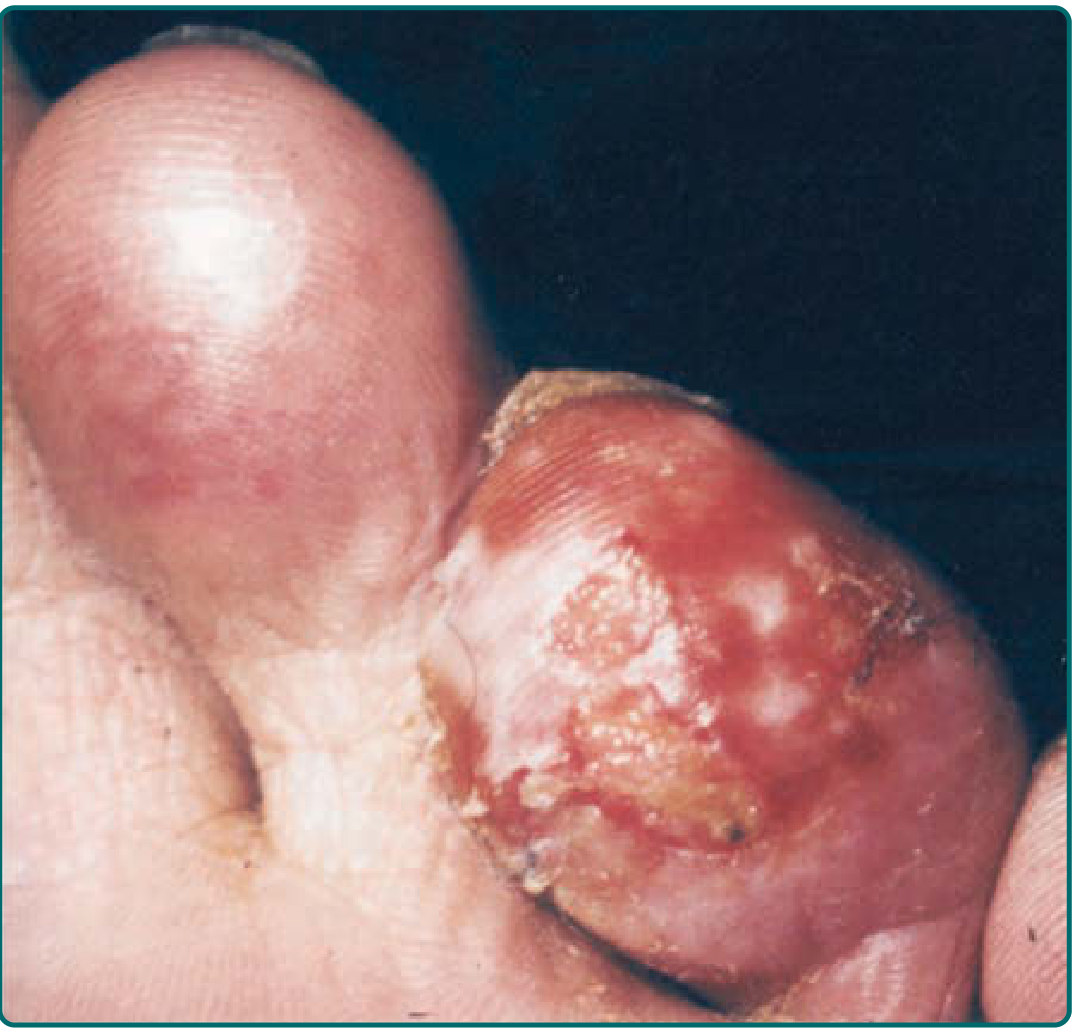
3d. Blister/Bulla Formation
- Subepidermal blisters form within 24-36 hours after rewarming
- Clear/white blebs = superficial injury, serous exudate (better prognosis)
- Hemorrhagic blebs = deep injury, damaged deeper vessels (poor prognosis)
- Blister fluid begins resorption within 5-10 days
Stage 4 - Extension of Injury (Progressive Ischemia)
- Platelet and leukocyte aggregation spreads into bordering viable tissue
- Microvascular thrombosis extends into previously uninjured zones
- Progressive ischemic necrosis occurs beyond the original freeze zone
- The injury "declares" itself wider than initially apparent
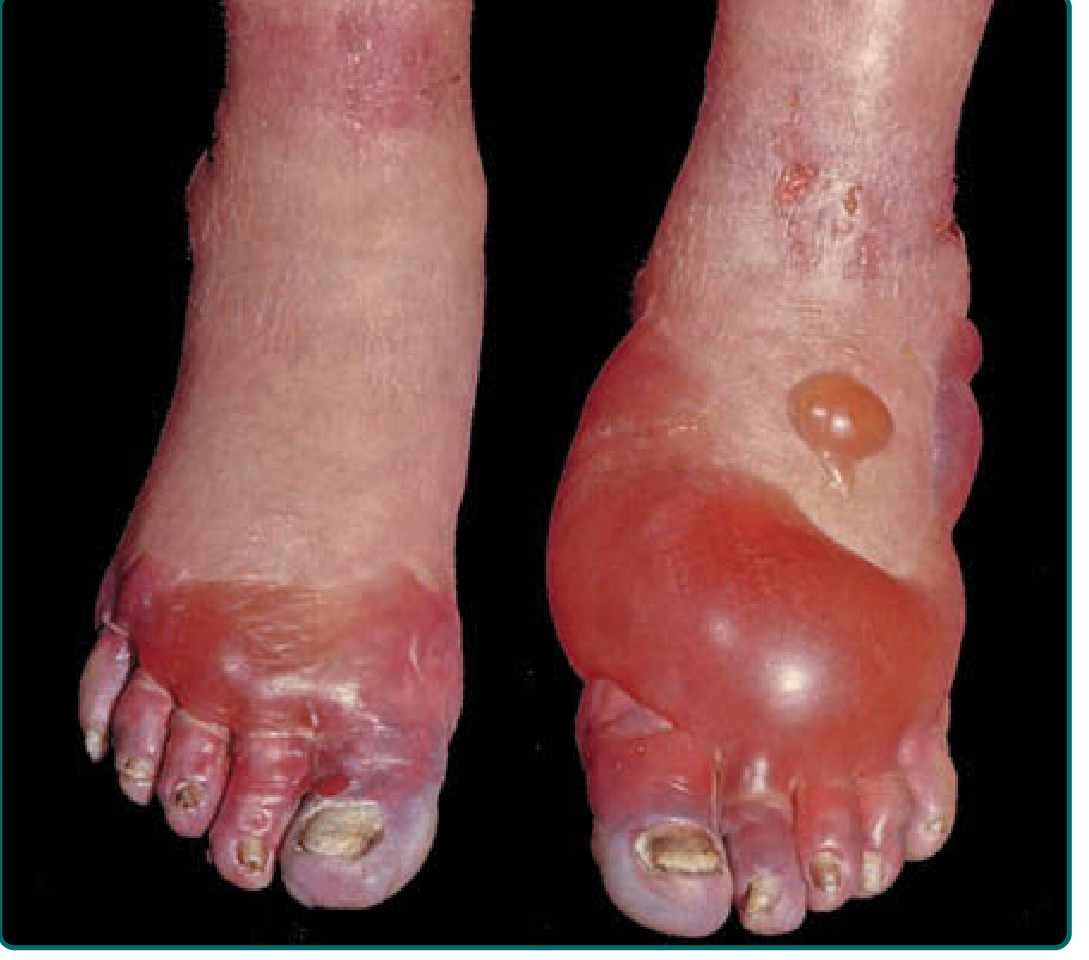
Stage 5 - Resolution or Necrosis
- Favorable outcome: Re-epithelialization, tissue recovery
- Unfavorable outcome: Eschar formation → hard, black dry gangrene (mummification)
- Over weeks, a line of demarcation forms
- Tissues distal to the line undergo autoamputation
- Vasomotor instability (cold sensitivity, hyperhidrosis, pain) may persist indefinitely - a consequence of lasting autonomic nerve injury
Summary Table - Four Physiologic Stages
| Stage | Key Events | Clinical Findings |
|---|---|---|
| I - Cooling | Hunting response fails; sustained vasoconstriction; extracellular then intracellular ice formation; erythrocyte sludging | Cool extremity, blanched, numb, woody texture |
| II - Thawing | Ice melts; cells swell; vessel wall failure; edema; inflammatory mediator release; platelet/leukocyte aggregation; thrombosis | Erythematous/violaceous, intense pain, blisters form |
| III - Extension | TXA2 surge; thrombosis extends into border tissue; vascular compromise widens | Blisters rupture |
| IV - Resolution | Re-epithelialization OR desiccation and mummification; persistent vasomotor instability | Healing or autoamputation |
Key Pathophysiologic Points to Remember
- Ice crystals form extracellularly first - intracellular formation requires very rapid freezing
- Rewarming is paradoxically the most damaging phase - reperfusion injury + inflammatory mediator peak
- TXA2 and PGF2α in blister fluid are the primary drivers of progressive tissue injury
- Freeze-thaw-refreeze cycles are catastrophic - each rewarming event spikes mediator levels further; field rewarming is contraindicated unless refreezing can be absolutely prevented
- Neuronal injury is particularly susceptible - peripheral nerves suffer at temperatures that spare other tissues, explaining persistent cold sensitivity and sensory neuropathy
Recent evidence note: A 2024 Wilderness Medical Society Practice Guideline (PMID: 38577729) and a 2026 review in Mil Med Res (PMID: 41953050) on frostbite diagnosis, treatment, and prognosis are the latest references - neither contradicts the above pathophysiology but may refine management recommendations.
Create a visual timeline diagram of the four pathophysiologic stages
A detailed medical educational timeline diagram showing the four pathophysiologic stages of frostbite, arranged horizontally left to right with arrows connecting each stage. Clean clinical infographic style on a dark navy blue background with white text. Stage 1 - COOLING (PRE-FREEZE): Ice blue color theme. Icon of an arrow pointing down (temperature dropping). Key events listed: "Hunting Reaction of Lewis", "Cyclic vasoconstriction & vasodilation", "Vasoconstriction becomes permanent", "Blood viscosity increases", "Tissue shifts to ambient temperature". Clinical signs: "Pallor, numbness, cold extremity, woody texture" Arrow pointing right → Stage 2 - FREEZING: Deep blue/white color theme. Snowflake icon. Key events: "Extracellular ice crystals form at -2°C", "Osmotic gradient dehydrates cells", "Cell membrane lysis", "Intravascular ice → erythrocyte sludging", "Microcirculatory flow ceases". Clinical signs: "Waxy white skin, anesthesia, painless, immobile joints" Arrow pointing right → Stage 3 - THAWING & REPERFUSION (most damaging): Red/orange color theme. Warning triangle icon. Key events: "Ice melts → reperfusion injury", "ROS generation → oxidative damage", "TXA2 & PGF2α surge in blister fluid", "Platelet & leukocyte aggregation", "Progressive microvascular thrombosis", "Blister/bulla formation (24–36 hrs)". Clinical signs: "Intense pain, erythema, edema, blisters" Arrow pointing right → Stage 4 - RESOLUTION OR NECROSIS: Dark brown/black color theme. Split into two outcomes with a fork: upper path shows green "Re-epithelialization → healing", lower path shows red "Mummification → dry gangrene → autoamputation". Key events: "Line of demarcation forms", "Vasomotor instability persists". Clinical signs: "Eschar, tissue loss, or recovery" At the bottom, a horizontal temperature scale bar from -2°C to body temperature. A note box at bottom: "Critical: Freeze-Thaw-Refreeze cycles are catastrophic — spike TXA2 further". The diagram title at top: "FROSTBITE: Stepwise Pathophysiology" in bold white text. Professional medical infographic, clear labels, anatomical accuracy.
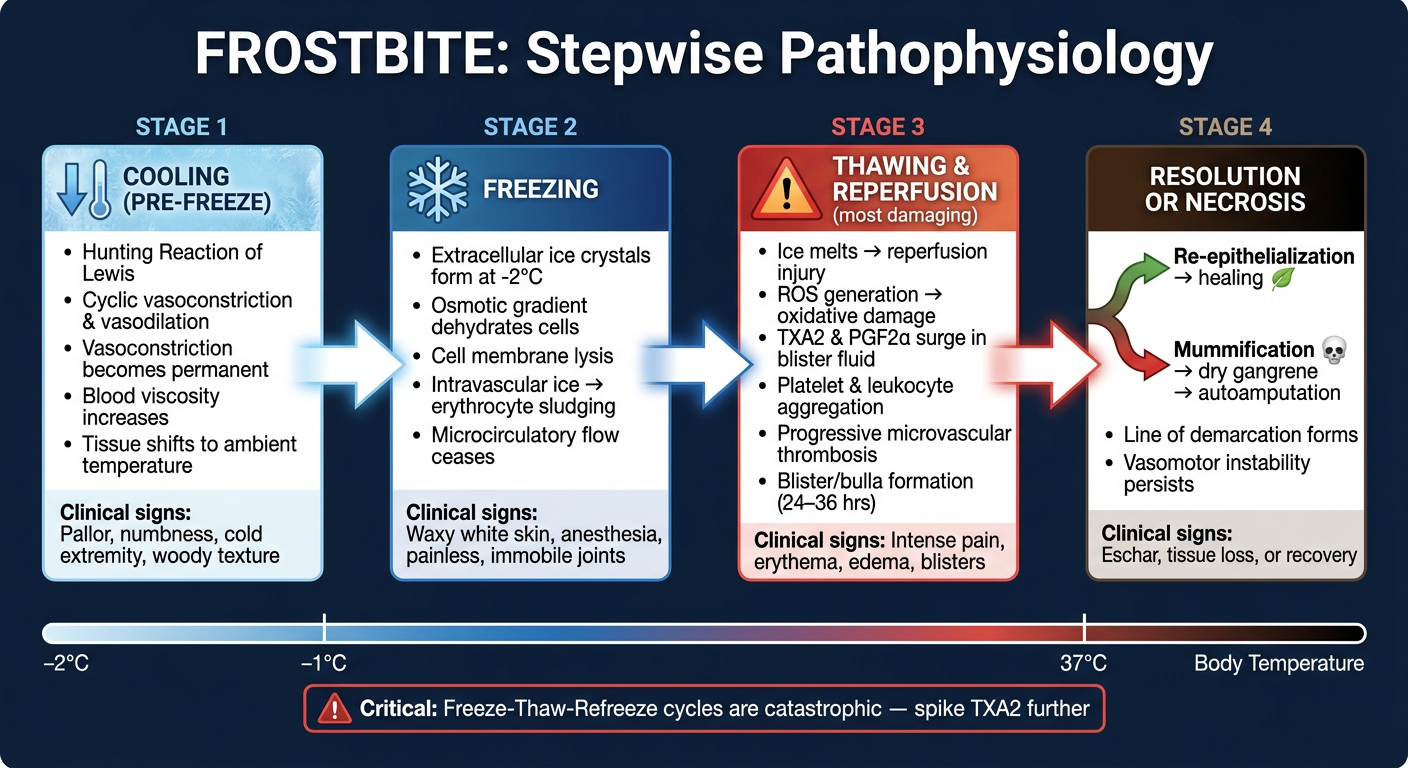
- Stage 1 (Cooling - Ice Blue): The Hunting Reaction of Lewis, sustained vasoconstriction, and rising blood viscosity
- Stage 2 (Freezing - Deep Blue): Ice crystal formation at -2°C, osmotic cell dehydration, membrane lysis, microvascular occlusion
- Stage 3 (Thawing/Reperfusion - Red/Orange): The most damaging phase - ROS generation, TXA2 + PGF2α surge, platelet/leukocyte aggregation, bulla formation
- Stage 4 (Resolution - Forked): Either re-epithelialization and healing (green path) or mummification, dry gangrene, and autoamputation (dark path)
How does tissue damage initiates inflammation?
How Tissue Damage Initiates Inflammation
Step 1 - Release of DAMPs (Damage-Associated Molecular Patterns)
| DAMP | Source |
|---|---|
| HMGB1 (High Mobility Group Box 1) | Nucleus - released from necrotic cells |
| Heat shock proteins (HSP60, HSP70, Gp96) | Cytoplasm |
| S100 proteins | Cytoplasm of neutrophils/macrophages |
| Uric acid / urate crystals | Purine metabolism breakdown |
| Heparan sulfate fragments | Degraded extracellular matrix |
| Hyaluronan oligomers | Degraded connective tissue |
| Fibronectin (extra domain A) | Disrupted ECM |
| Mitochondrial DNA / fragments | Released from damaged mitochondria |
| IL-1α | Constitutively present in cell cytoplasm |
These molecules are not normally present in the extracellular space - their presence signals structural cell death to the immune system.
Step 2 - PRR Recognition (Pattern Recognition Receptors)
- Toll-Like Receptors (TLRs) - membrane-bound; recognize extracellular DAMPs
- NOD-Like Receptors (NLRs / nucleotide-binding leucine-rich repeat receptors) - intracellular; sense intracellular stress signals
- RAGE (Receptor for Advanced Glycation End-products) - binds HMGB1 and S100 proteins
- Purinergic receptors (P2X7) - detect extracellular ATP released from dying cells
Step 3 - Sentinel Cell Activation (Tissue Macrophages & Mast Cells)
- They are already present in all tissues, sampling the environment continuously
- Upon DAMP/PRR activation, they immediately release a burst of pre-formed and newly synthesized mediators:
| Mediator | Effect |
|---|---|
| Histamine | Rapid vasodilation + increased vascular permeability |
| Eicosanoids (prostaglandins, leukotrienes) | Vasodilation, pain, fever |
| Tryptases | Activate more mast cells (amplification) |
| TNF-α | Early cytokine; induces endothelial adhesion molecules |
| IL-1β | Activates endothelium; triggers fever; activates more cells |
| Chemokines | Chemoattractants to recruit circulating leukocytes |
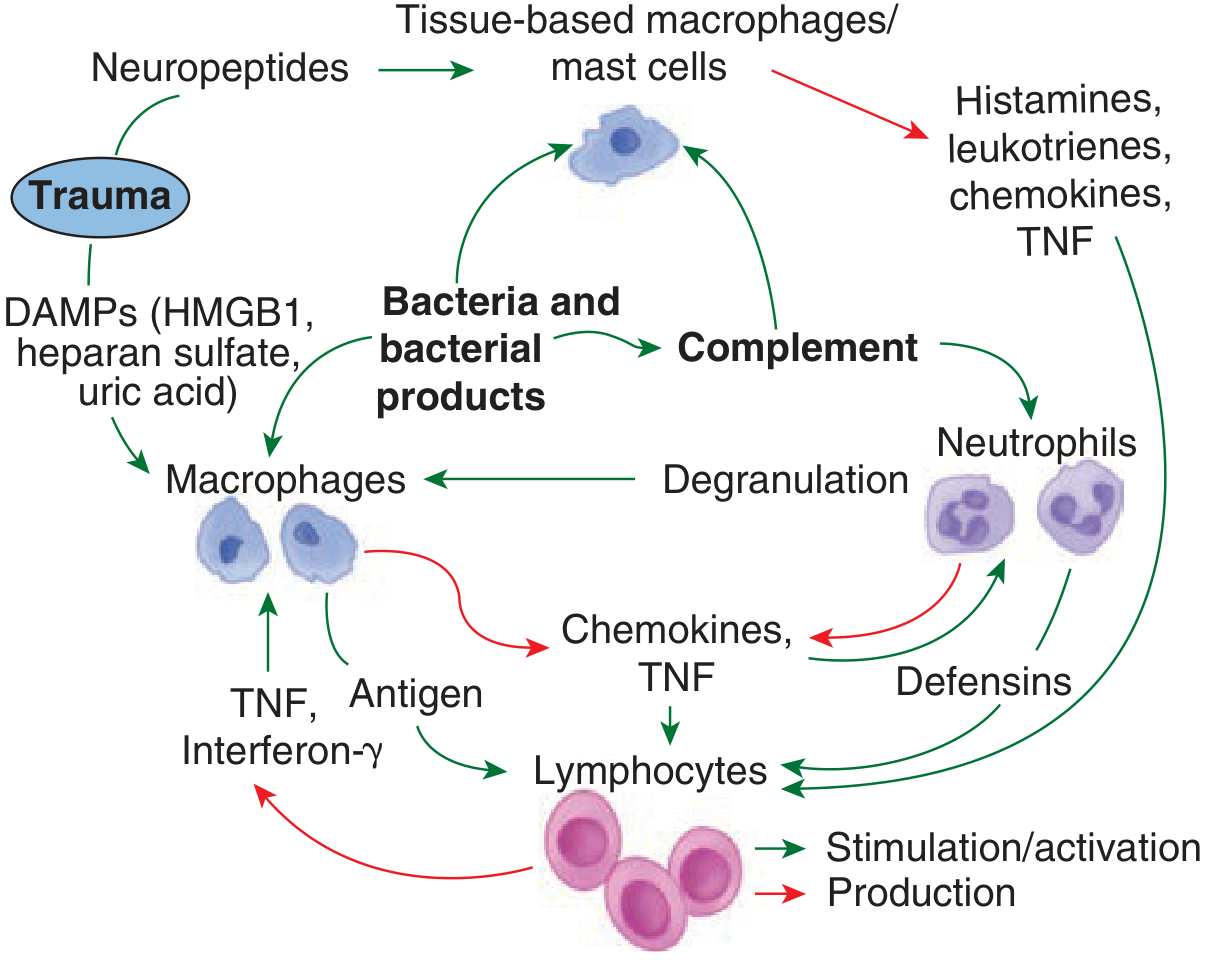
Step 4 - Inflammasome Assembly
- Caspase-1 is activated within the inflammasome
- Active caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature active forms
- This dramatically amplifies the local inflammatory signal
- Inflammasome activation also triggers pyroptosis - a highly inflammatory form of programmed cell death that releases more DAMPs, creating a self-amplifying loop
Inflammasomes can also be activated in endothelial cells and platelets, directly causing leaky capillaries and coagulopathy - and the resulting local ischemia generates yet more DAMPs.
Step 5 - Vascular Changes (The Cardinal Signs Explained)
- Vasodilation → increased blood flow → rubor (redness) and calor (heat)
- Increased vascular permeability → plasma leaks into interstitium → tumor (swelling)
- Compression of sensory nerves + bradykinin → dolor (pain)
- E-selectin expressed within 2 hours → mediates neutrophil rolling
- P-selectin expressed rapidly (from Weibel-Palade bodies) → early neutrophil capture
- VCAM-1 expressed at 6-12 hours → recruits monocytes and lymphocytes
- ICAM-1 and IL-8 displayed on endothelial surface → firm adhesion and chemotaxis
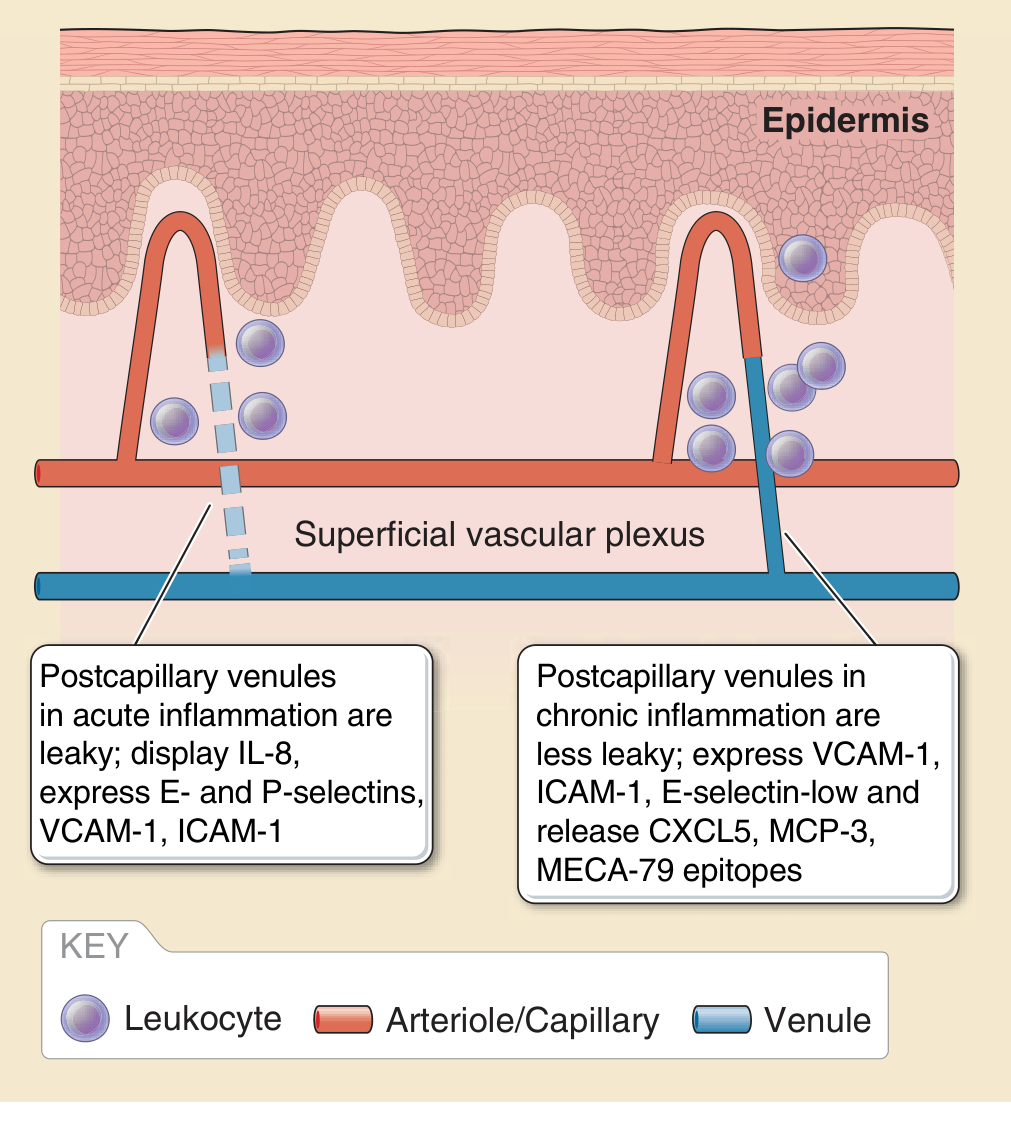
Step 6 - Leukocyte Recruitment Cascade
- Rolling - weak, reversible binding via selectins (E-selectin/P-selectin on endothelium bind PSGL-1 on leukocytes)
- Activation - chemokines (IL-8/CXCL8) on the endothelial surface activate integrin affinity
- Firm adhesion - integrins (LFA-1, Mac-1) bind ICAM-1 → leukocyte stops
- Transmigration (diapedesis) - leukocyte squeezes between endothelial cells through tight junction gaps opened by histamine and cytokines
- Chemotaxis - leukocyte migrates along the DAMP/chemokine gradient to the injury site
- Neutrophils first (within 30-60 minutes; peak 6-12 hours) - phagocytosis, ROS generation
- Monocytes/Macrophages follow (12-24 hours) - phagocytosis, orchestration of repair
- Lymphocytes later (days) - adaptive immune response if needed
Step 7 - Amplification and Systemic Spread
- TNF-α (peaks at 90 min): peripheral vasodilation, activates more cytokines, induces procoagulant activity
- IL-6: hepatic acute-phase response → CRP, fibrinogen, serum amyloid A synthesis
- IL-1β + TNF → hypothalamus → fever (via PGE2)
- Complement activation (alternative and classical pathways) → C3a and C5a (anaphylatoxins) → mast cell degranulation, neutrophil chemotaxis
- Coagulation/kinin cascades activated → bradykinin, thrombin → further amplification
Step 8 - Self-Amplification and Secondary Triggers
"Triggers to further release of DAMPs include sepsis, haemorrhage, massive transfusion, acidosis, surgery, crush syndrome and ischaemia-reperfusion. Thus secondary insults of delayed or ineffective treatment... will tend to maintain and amplify the inflammatory process." - Bailey & Love's Surgery
Summary Flowchart
TISSUE INJURY
↓
[Cell necrosis / membrane disruption]
↓
DAMPs released (HMGB1, HSPs, S100, uric acid, mtDNA, ATP)
↓
PRRs activated (TLRs, NLRs, RAGE) on macrophages & mast cells
↓
NF-κB signaling → transcription of pro-inflammatory genes
↓
Inflammasome assembly (NLRP3) → caspase-1 activation → IL-1β, IL-18 maturation
↓
Sentinel cells release: Histamine, TNF-α, IL-1, IL-6, chemokines, eicosanoids
↓
Endothelial activation → E/P-selectin, VCAM-1, ICAM-1 expression
↓
Vascular dilation + increased permeability (rubor, calor, tumor, dolor)
↓
Leukocyte rolling → adhesion → diapedesis → chemotaxis to injury site
↓
Neutrophil/macrophage phagocytosis + ROS + more mediator release
↓
[Controlled] → Resolution & repair
[Uncontrolled] → SIRS → organ dysfunction → MODS
Why there is no pain in stage 2 of frostbite and intense pain in stage 3
Why No Pain in Stage 2 (Freezing) but Intense Pain in Stage 3 (Rewarming)?
Stage 2 (Freezing) - Why There is NO Pain
1. Cold-Induced Failure of Nerve Conduction
- At ~15°C, cold thermoreceptors stop firing (paradoxically - they fire maximally around 25°C, then are silenced by further cold)
- At ~7-10°C, nerve conduction velocity slows dramatically as membrane fluidity decreases and ion channel kinetics slow
- Below ~4-5°C, voltage-gated sodium channels (Nav) become functionally inactivated - the channel gates are physically slowed by cold, so action potentials cannot propagate
- At tissue-freezing temperatures (~-2°C and below), conduction block is complete - no electrical signal can travel along the nerve
2. Ischemic Neuropraxia
- Sustained vasoconstriction (from the failed hunting reaction) drastically reduces blood flow to the nerve
- Nerves are among the most metabolically sensitive tissues - they require continuous oxygen and glucose for Na-K-ATPase activity to maintain the resting membrane potential
- Ischemia depletes ATP → Na-K-ATPase fails → the nerve depolarizes and becomes inexcitable (cannot fire)
- The axon enters a state of neuropraxia - temporary conduction failure without structural axon damage (at this point, reversible)
3. Ice Crystal Compression of Nerve Microarchitecture
- Extracellular ice crystal formation compresses the endoneurial space
- Physical distortion of the axon membrane prevents normal receptor-to-cortex signal transmission
- The nerve terminals themselves are frozen - transducer proteins in nociceptors cannot open their ion channels when lipid membranes are crystallized
Stage 3 (Thawing/Rewarming) - Why There is INTENSE Pain
1. Restoration of Nerve Conduction - The Nociceptors "Wake Up"
- As temperature rises above 10°C, Nav channel kinetics normalize
- Nerve conduction resumes - first in larger, faster Aβ fibers, then in Aδ and C fibers
- The nociceptors (both TRPA1 cold/pain receptors and TRPV1 heat-pain receptors) become functional again
- Now they have a massive amount of tissue damage to report
2. Ischemia-Reperfusion Re-Activates and Sensitizes Nociceptors
- Reactive oxygen species (ROS) are generated - these directly activate TRPA1 channels on C-fibers, producing burning pain
- Extracellular acidosis (from accumulated lactate during the ischemic period) activates ASIC (acid-sensing ion channels) on nociceptors
- ATP released from damaged cells activates P2X3 purinergic receptors on sensory nerve terminals
3. Inflammatory Mediator Surge - "Chemical Soup" of Sensitization
| Mediator | Effect on Nociceptors |
|---|---|
| Prostaglandin E2 (PGE2) / PGF2α | Directly sensitizes Aδ and C fibers via EP receptors - lowers the firing threshold (peripheral sensitization) |
| Thromboxane A2 (TXA2) | Vasoconstriction + direct nociceptor sensitization |
| Bradykinin | Powerfully activates B1/B2 receptors on C-fibers - one of the most potent pain-inducing mediators known |
| Histamine | Activates H1 receptors on C-fibers → itch and burning pain |
| IL-1β and TNF-α | Upregulate Nav1.7 and Nav1.8 sodium channel expression → lowers action potential threshold |
| Substance P and CGRP | Released from activated C-fibers themselves → neurogenic inflammation, vasodilation, further sensitization |
4. Edema Compresses Nerve Terminals
- Vascular permeability increases massively during rewarming
- Tissue edema within tight fascial compartments creates mechanical pressure on nerve terminals and free nerve endings
- This adds a pressure-nociception component to the already chemically-sensitized pain
Summary Comparison
| Feature | Stage 2 (Freezing) | Stage 3 (Rewarming) |
|---|---|---|
| Temperature | ~-2°C and below | Rising to 37-42°C |
| Nav channel state | Inactivated by cold - no action potentials | Fully functional |
| Blood flow | Near zero (ischemic neuropraxia) | Restored - reperfusion |
| Nociceptor state | Physically frozen, conduction blocked | Active + peripherally sensitized |
| Prostaglandins/TXA2 | Low (synthesis requires enzymatic activity, impaired by cold) | Peak concentration |
| Bradykinin/Histamine | Minimal | Massively elevated |
| ROS | None (no reperfusion yet) | Generated on reoxygenation |
| Clinical pain | None - complete anesthesia | Intense burning, throbbing pain |
Clinical Implication
Step wise pathophysiology of deep burns
Stepwise Pathophysiology of Deep Burns
Step 1 - Direct Thermal Injury: Protein Denaturation and Coagulative Necrosis
- Cellular proteins denature - the hydrogen bonds stabilizing tertiary protein structure are broken by heat; enzyme systems fail, structural proteins collapse
- Cell membranes are disrupted - lipid bilayers are directly damaged; membrane permeability is lost
- Collagen and elastin are destroyed - the structural scaffold of the dermis becomes non-functional
- Nucleic acids are damaged - DNA strand breaks prevent any possibility of cellular repair
Step 2 - The Three Zones of Jackson (Local Wound Architecture)
Zone 1 - Zone of Coagulation (Center)
- Maximum temperature, maximum damage
- Irreversible coagulative necrosis - all cells dead
- Vessels are thrombosed, proteins coagulated
- This becomes the eschar (hard, leathery, insensate tissue)
- No recovery possible without surgical debridement
Zone 2 - Zone of Stasis (Middle)
- Cells are injured but potentially viable
- Microvascular damage causes sluggish flow, endothelial activation, and progressive thrombosis
- Without intervention, this zone converts to necrosis within 48-72 hours due to ischemia, edema, and inflammatory damage
- This is the clinically critical zone - the target of resuscitation and topical therapy
Zone 3 - Zone of Hyperemia (Periphery)
- Minimal cellular injury
- Vasodilation and increased perfusion (classic inflammatory response)
- Fully reversible - heals spontaneously within 7-10 days
Key clinical point: The goal of burn resuscitation is to salvage the Zone of Stasis - failure to do so converts a deep partial-thickness burn into a full-thickness one.
Step 3 - Cell Death Mechanisms Within the Wound
| Mechanism | Trigger | Inflammatory Signal |
|---|---|---|
| Necrosis / Necroptosis | Direct thermal destruction | Massive DAMP release (HMGB1, HSPs, uric acid) → intense inflammation |
| Apoptosis | Sub-lethal heat, ROS, ischemia at wound edges | Contained cell death, less inflammation |
| Autophagy | Cellular stress response | Potentially protective - may limit injury propagation |
Step 4 - Local Inflammatory Response (Minutes to Hours)
- Histamine - released from dermal mast cells within seconds; causes immediate vasodilation and increased vascular permeability
- Serotonin - vasoactive amine released from platelets activated by exposed collagen
- Prostaglandins and leukotrienes - synthesized within minutes; sustain vasodilation, attract neutrophils
- Thromboxane A2 - causes vasoconstriction in the zone of stasis microcirculation → ischemia → contributes to zone conversion
- Reactive oxygen species (ROS) - generated by ischemia-reperfusion at wound edges; direct cell membrane damage
- Cytokines (TNF-α, IL-1β, IL-6, IL-8) - amplify inflammation, activate endothelium
- E-selectin and P-selectin expressed (neutrophil rolling)
- VCAM-1 and ICAM-1 upregulated (firm adhesion)
- Tight junctions open → massive increase in capillary permeability
- Plasma proteins and fluid pour into the interstitium → burn edema
- First peak: within 1 hour (histamine, complement, immediate mediators)
- Second peak: 12-24 hours (cytokine-driven, neutrophil-mediated)
Step 5 - Burn Edema and Fluid Compartment Shifts
- Massive capillary leak (from histamine, ROS, bradykinin, IL-1) → loss of plasma proteins into the interstitium
- Loss of oncotic pressure gradient (hypoproteinemia) - the main force retaining fluid in vessels is abolished
- Hydrostatic pressure now exceeds oncotic pressure → fluid pours into interstitium (Starling forces imbalance)
- In burns >20% TBSA, this is not confined to the wound - capillary leak occurs systemically through non-burned tissue as well (circulating mediators damage vessels throughout the body)
- Massive intravascular volume depletion → burn shock (hypodynamic "ebb phase")
- Decreased cardiac output within the first 24-72 hours
- Hemoconcentration (raised hematocrit)
- Tissue hypoperfusion → lactic acidosis
- Risk of acute kidney injury (from hypovolemia + direct myoglobin toxicity)
- Phase 1 (0-24h): "Ebb phase" - capillary leak, decreased cardiac output, increased systemic vascular resistance
- Phase 2 (24-72h): "Flow phase" - vascular permeability normalizes, fluid begins reabsorption, cardiac output increases (often to hyperdynamic levels)
"Imbalance between oncotic and hydrostatic forces develops, making resuscitation essential at this stage." - Rosen's
Step 6 - Loss of Skin Barrier Function
| Lost Function | Consequence |
|---|---|
| Microbial barrier | Bacterial colonization of eschar begins within 24-48h; sepsis risk |
| Epidermal water barrier (stratum corneum/granulosum) | Insensible fluid losses of up to 3-5 L/day/m² of burn - far exceeding normal |
| Thermoregulation (sweat glands, dermal plexus) | Inability to regulate body temperature; hypothermia risk intraoperatively |
| Sensory function (mechanoreceptors, nociceptors) | Permanent sensory loss in full-thickness burns |
| Elasticity (elastin) | Rigid eschar restricts movement; circumferential burns compress limbs/chest |
Step 7 - Eschar Formation and Secondary Wound Events
- Devitalized, leathery, non-viable tissue
- Initially sterile, but within 3-7 days, bacterial colonization begins from the patient's own skin flora and gut translocation
- Bacteria produce toxins → stimulate further local and systemic inflammation
- In burns >40% TBSA, bacterial load becomes so overwhelming that without debridement, sepsis and cardiovascular collapse are inevitable
- The dead eschar is also a continuous source of DAMPs - perpetually stimulating the inflammatory response
- Rigid eschar around limbs → compartment syndrome → ischemic necrosis of muscle and nerve
- Rigid eschar around chest → respiratory failure (prevents rib expansion)
- Treatment: escharotomy (surgical incision of eschar)
Step 8 - Systemic Inflammatory Response Syndrome (SIRS)
- Fever (IL-1β, IL-6, TNF-α act on hypothalamic PGE2 production)
- Hyperdynamic circulation - heart rate and cardiac output increase dramatically (flow phase, from 24-72h onward)
- Diffuse capillary leak in non-burned tissues - pulmonary edema, cerebral edema
- Immunosuppression - paradoxically, sustained SIRS eventually suppresses immune effector function (compensatory anti-inflammatory response syndrome, CARS), increasing susceptibility to infections
Step 9 - Hypermetabolic Response (Days to Weeks)
- Hypothalamic-pituitary-adrenal (HPA) axis activated → cortisol surge → insulin resistance, protein catabolism
- Sympathoadrenal activation → catecholamine surge → tachycardia, hyperglycemia, lipolysis
- Metabolic rate rises 2-3× above normal (greatest hypermetabolism of any injury)
- Muscle proteolysis - structural proteins degraded to amino acids; glutamine stores depleted to 50% of normal; muscle wasting can be profound
- Amino acids feed hepatic gluconeogenesis and acute-phase protein synthesis (CRP, fibrinogen, α2-macroglobulin, complement)
- Peripheral lipolysis → free fatty acids → hepatic oxidation or reesterification → fatty liver development
- Hyperglycemia (from gluconeogenesis + insulin resistance) → worsens outcomes
Step 10 - Wound Healing Phases (or Failure)
| Phase | Timing | Key Events |
|---|---|---|
| Inflammatory | 0-4 days | Neutrophil infiltration, debridement of necrotic tissue, macrophage takeover |
| Proliferative | 4 days - 3 weeks | Angiogenesis, fibroblast migration, granulation tissue formation, myofibroblast contraction |
| Remodeling | Weeks to years | Collagen remodeling - irregular bundles (not parallel), fibrosis, contracture formation |
- Hypertrophic scarring - disordered collagen bundles with only 80% tensile strength
- Contractures - myofibroblast contraction of scar tissue across joints → functional limitation
- Marjolin's ulcer - squamous cell carcinoma developing in chronic burn scars (melanin-deficient scar lacks UV protection)
Summary Timeline
HEAT EXPOSURE
↓
[Protein denaturation + coagulative necrosis — Seconds]
↓
Three Zones: Coagulation / Stasis / Hyperemia
↓
[DAMPs released → Mast cell/Macrophage activation — Minutes]
Histamine, TXA2, PGE2, ROS, Cytokines
↓
[Endothelial activation + Capillary leak — Minutes to Hours]
Massive edema, Hypoproteinemia, Intravascular volume loss
↓
[BURN SHOCK (Ebb phase) — 0-24h]
↓ Cardiac output, ↑ SVR, Tissue hypoperfusion, AKI risk
↓
[Zone of Stasis converts to necrosis without resuscitation — 48-72h]
↓
[SIRS + Loss of barrier function — Hours to Days]
Fever, Fluid shifts, Immune activation, Bacterial colonization
↓
[Flow/Hypermetabolic Phase — 24h to months/years]
↑↑ Cardiac output, ↑↑ Metabolic rate (2-3×), Catabolism, Insulin resistance
↓
[Wound healing (if survives) — Weeks to Years]
Scar, Contracture, Marjolin's ulcer risk