Presentation , investigation and management of MDS
"myelodysplastic syndrome"[MeSH Terms] AND management
"myelodysplastic syndrome"[MeSH Terms] AND treatment
"myelodysplastic syndromes"[MeSH Terms] AND diagnosis
Myelodysplastic Syndromes (MDS)
Definition
- Ineffective hematopoiesis leading to peripheral blood cytopenias
- Dysplastic maturation of one or more myeloid cell lines
- High risk of transformation to AML (in 10-40% of cases)
Epidemiology & Risk Factors
- Predominantly a disease of older adults (mean age at onset ~70 years); median age at diagnosis is in the seventh to eighth decade
- Affects approximately 15,000 people per year in the US - about as common as AML
- Slight male predominance
-
Environmental: radiation, benzene, smoking
-
Therapy-related (t-MDS): alkylating agents (busulfan, nitrosourea, procarbazine) - latency 5-7 years; topoisomerase II inhibitors - latency 2 years
-
Preceding hematologic disease: acquired aplastic anemia, Fanconi anemia, paroxysmal nocturnal hemoglobinuria (PNH)
-
Constitutional/germline mutations: GATA2 (MonoMAC syndrome), RUNX1, telomere gene mutations (consider in patients <40 years)
-
Clonal hematopoiesis of indeterminate prognosis (CHIP) - common precursor, progresses to MDS at ~1% per year
-
Harrison's 22E, p. 859-860
Pathogenesis
| Category | Examples | Clinical Significance |
|---|---|---|
| Epigenetic factors | TET2, DNMT3A, EZH2, IDH1/2, ASXL1 | DNA methylation and histone modification dysregulation |
| RNA splicing factors | SF3B1, SRSF2, U2AF1 | SF3B1 mutations strongly associated with ring sideroblasts and favorable prognosis |
| Transcription factors | RUNX1, GATA2 | Loss-of-function mutations; deranged myeloid differentiation |
Presentation (Clinical Features)
Symptoms
- Anemia dominates the early course: fatigue, weakness, dyspnea, pallor (most common presentation)
- At least half of patients are asymptomatic, discovered incidentally on routine blood counts
- Infections from neutropenia (functional and/or quantitative)
- Bleeding/bruising from thrombocytopenia or platelet dysfunction
- Fever and weight loss are more suggestive of a myeloproliferative rather than myelodysplastic process
Physical Examination
-
Signs of anemia (pallor, tachycardia)
-
~20% of patients have splenomegaly
-
Associated autoimmune manifestations (Sweet's syndrome, arthritis)
-
VEXAS syndrome should be considered when MDS coexists with inflammatory disease (UBA1 somatic mutation)
-
Constitutional anomalies (short stature, abnormal thumbs) point to Fanconi anemia in younger patients
-
Harrison's 22E, p. 860
Classification
ICC (International Consensus Classification, 2022) Key Subtypes:
| Subtype | Blasts | Key Features |
|---|---|---|
| MDS with 5q deletion (MDS-5q) | <5% BM, <2% PB | Isolated 5q deletion; favorable prognosis |
| MDS with SF3B1 mutation (MDS-SF3B1) | <5% BM, <2% PB | Ring sideroblasts; favorable prognosis |
| MDS with biallelic TP53 (MDS-biTP53) | Any | Complex karyotype; very poor prognosis |
| MDS with excess blasts (MDS-EB) | 5-19% BM | Higher risk of AML transformation |
| MDS/AML | 10-19% BM | New category emphasizing disease continuum |
- Harrison's 22E, p. 859
Investigations
1. Full Blood Count and Peripheral Blood Smear
- Anemia (usually macrocytic; can be normocytic or microcytic) - present in most cases
- Thrombocytopenia - large, hypogranular platelets with functional defects
- Neutropenia - neutrophils are hypogranulated, show hyposegmented nuclei (Pseudo-Pelger-Hüet cells), contain Döhle bodies
- Circulating myeloblasts (correlate with marrow blast count)
- Total WBC usually normal or low (except in CMML where monocytosis is prominent)
- Elevated RDW, poikilocytes, macroovalocytes
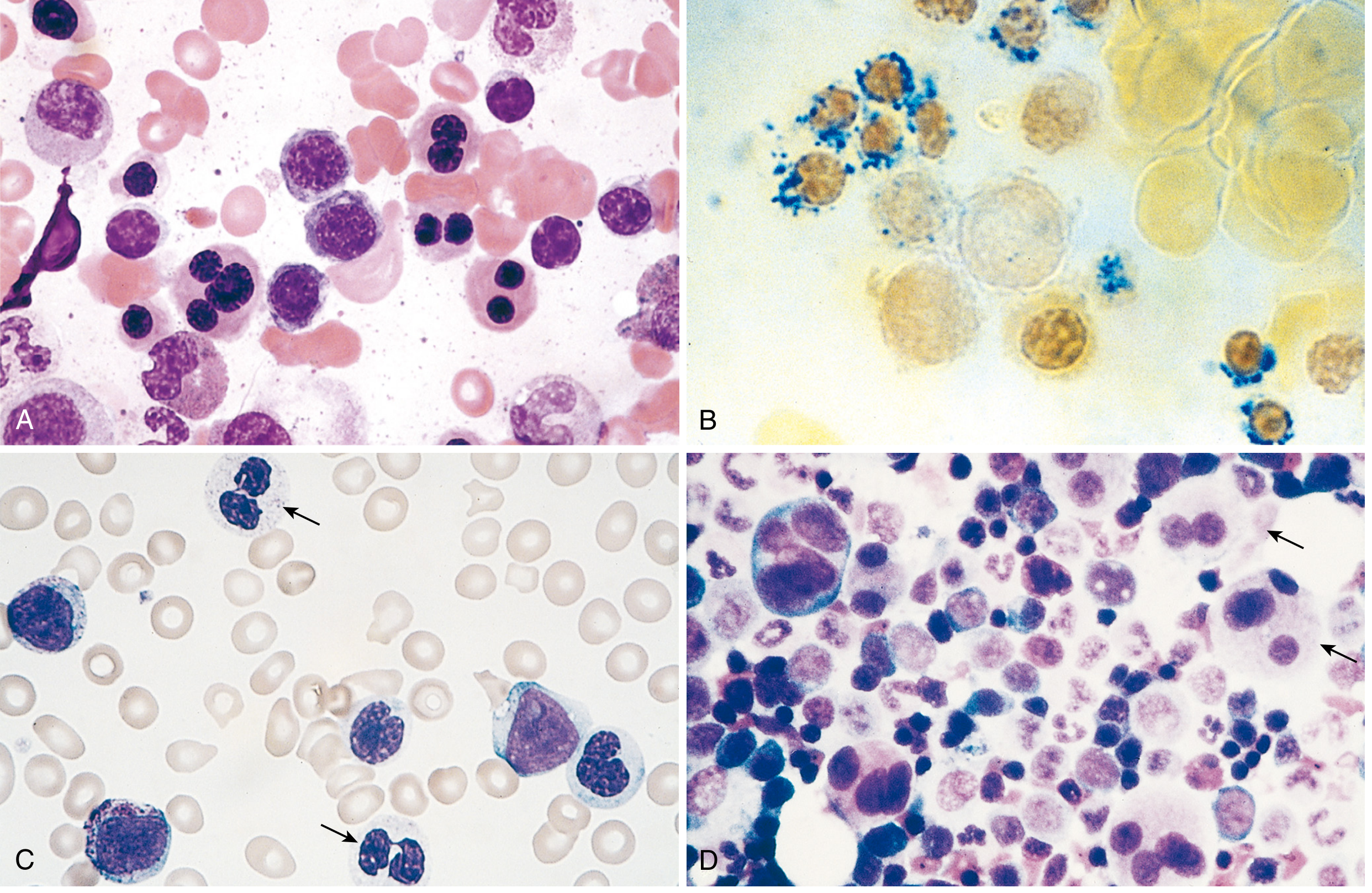
2. Bone Marrow Aspirate and Trephine Biopsy (Mandatory)
- Ring sideroblasts - erythroblasts with iron-laden mitochondria visible as perinuclear granules on Prussian blue stain
- Megaloblastoid maturation
- Nuclear budding abnormalities (multilobated or multiple nuclei)
- Decreased secondary granules
- Pseudo-Pelger-Hüet cells (bilobed nuclei)
- Neutrophils without nuclear segmentation
- Döhle bodies, toxic granulations
- Small megakaryocytes with single small nuclei (micromegakaryocytes)
- Large megakaryocytes with multiple separate nuclei ("pawn-ball" megakaryocytes)
3. Cytogenetics (Conventional Karyotype + FISH)
- Conventional karyotype on bone marrow aspirate: identifies chromosomal abnormalities in ~50% of cases
- Common: del(5q), del(7q), monosomy 7, monosomy 5, trisomy 8, del(20q)
- Complex karyotype (>3 abnormalities) = very poor prognosis, predicts AML evolution
- Several cytogenetic abnormalities provide presumptive evidence of MDS even when morphologic dysplasia is insufficient
- Copy number abnormalities detected by array CGH/SNP array
4. Molecular/Genomic Testing (Next-Generation Sequencing)
- Now routinely performed in MDS workup
- Detects mutations in SF3B1, TET2, DNMT3A, ASXL1, RUNX1, EZH2, TP53, SRSF2, U2AF1, IDH1/2
- Essential for ICC/WHO 2022 classification (molecularly defined subtypes)
- Prognostic IPSS-M (2022) incorporates molecular data
- Distinguish pathogenic mutations from CHIP and variants of uncertain significance
5. Additional Laboratory Tests
- Serum B12 and folate - to exclude megaloblastic anemia (essential differential)
- Serum vitamin B6 - trial of pyridoxine if ring sideroblasts present
- Copper levels - copper deficiency can mimic MDS
- Serum ferritin, LDH, β2-microglobulin - for IPSS-R scoring
- Serum EPO - important for treatment decisions (lower EPO predicts better response to ESAs)
- Flow cytometry - reveals aberrant hematopoietic differentiation; enumerates blasts; detects PNH clone
- Iron studies - assess iron overload from transfusions
- Thyroid function - in macrocytic anemia workup
6. Differential Diagnosis to Exclude
- Vitamin B12/folate deficiency
- Copper deficiency
- Hypothyroidism
- Drug effects (methotrexate, hydroxycarbamide, alcohol)
- Aplastic anemia (hypocellular MDS vs. aplastic anemia)
- PNH
- HIV infection
- Congenital dyserythropoietic anemias (in younger patients)
Prognosis: IPSS-R Scoring
- Marrow blast count categories (≤2%, >2-<5%, 5-10%, >10-30%)
- Cytogenetic risk group (5 subgroups, 16 specific abnormalities)
- Depth of cytopenias (Hb, neutrophil count, platelet count)
- Age, performance status, serum ferritin, LDH, β2-microglobulin
- Very low / Low risk: years (up to >5 years for MDS-5q)
- Intermediate risk: ~3 years
- High / Very high risk: months (as low as <6 months for excess blasts with complex karyotype)
Management
A. Low-Risk MDS (IPSS-R Low/Very Low/Intermediate)
- RBC transfusions for symptomatic anemia; accompanied by iron chelation (deferoxamine or deferasirox) to prevent secondary hemochromatosis
- Platelet transfusions for bleeding
- Antibiotic prophylaxis and prompt treatment of infections
- G-CSF for severe neutropenia
- EPO alone or with G-CSF improves Hb levels, particularly in patients with low serum EPO levels and low transfusion requirements
- Survival improved by EPO through amelioration of anemia
- G-CSF alone did not improve survival in controlled trials
- Inhibits TGF-β-mediated suppression of erythropoiesis
- FDA approved for anemia in low-risk MDS, particularly those with SF3B1 mutations
- Reduces transfusion burden
- Particularly effective in del(5q) MDS: ~67% achieve transfusion independence
- Remarkably, cytogenetics also normalize in many patients
- Oral administration; response usually within 3 months
- Toxicities: worsening thrombocytopenia/neutropenia, DVT/PE risk
- For younger patients (<60 years) with hypocellular marrow and HLA-DR15 positivity
- Antithymocyte globulin (ATG) + cyclosporine: ~50% response
- Alemtuzumab (anti-CD52 monoclonal antibody) also used
- Mimics the immunosuppressive approach used in aplastic anemia
B. High-Risk MDS (IPSS-R High/Very High)
-
Azacitidine (5-azacytidine): 75 mg/m² SC for 7 days every 4 weeks
- Improves blood counts and survival vs. best supportive care
- ~50% of patients show improved blood counts/decreased transfusion requirements
- Requires minimum 4 cycles to assess response; continued administration needed
- Superior to decitabine in improving overall survival in randomized trials
-
Decitabine: 30-50% response rate, duration ~1 year; IV infusion over 3-10 days in repeating cycles
-
Oral decitabine-cedazuridine: FDA approved for high-risk MDS (oral formulation with bioavailability comparable to IV)
-
Only known curative treatment for MDS
-
Survival ~50% at 3 years in selected patients
-
Indications: younger patients, higher-risk IPSS-R, earlier in disease course
-
Reduced-intensity conditioning (RIC) allows transplantation in older patients with acceptable toxicity; slightly higher relapse risk
-
Unrelated matched donor results comparable to sibling donors
-
The "transplant conundrum": high-risk patients most need it but tolerate it worst; low-risk patients tolerate it best but may do well for years with less aggressive therapy
-
In reality, only a small proportion of MDS patients actually undergo transplantation
-
Goldman-Cecil Medicine, p. 1908
- Ivosidenib (IDH1 inhibitor): FDA approved for MDS with IDH1 mutations
- Venetoclax (BCL2 inhibitor): FDA approved for AML; used in combination with HMAs in high-risk MDS (investigational)
- Enasidenib (IDH2 inhibitor): used in IDH2-mutated MDS
- Considered for eligible patients as a bridge to transplant
- AML-type induction regimens (cytarabine + anthracycline) - responses are generally poor and not durable
- MDS is regarded as particularly refractory to cytotoxic regimens
C. Novel/Emerging Therapies (Trials)
- TP53 inhibitors (targeting TP53-mutant MDS)
- Splicing factor mutation-targeted agents
- Inflammasome inhibitors / IL-1 receptor-associated kinase (IRAK) inhibitors
- Imetelstat (telomerase inhibitor) - phase III trials underway
- HMA + venetoclax combinations
- Likely future: HMA monotherapy will be replaced by HMA combination regimens
Summary Algorithm
MDS diagnosed
|
Risk stratification (IPSS-R / IPSS-M)
|
Low/Intermediate risk High risk
| |
- Supportive care - Consider allo-HSCT
- ESAs (if low EPO) (if eligible: younger, fit,
- Luspatercept (SF3B1+) suitable donor)
- Lenalidomide (5q del) |
- Immunosuppression Not eligible for transplant
(hypocellular, young) |
- Hypomethylating agents
(azacitidine preferred)
- Targeted agents (IDH1/2 i)
- Clinical trials
- Supportive care
Key Points to Remember
- MDS is primarily a disease of the elderly (median age 70); cytopenias with hypercellular marrow is the paradox
- Bone marrow biopsy with cytogenetics and molecular testing is mandatory for diagnosis and classification
- Pseudo-Pelger-Hüet cells and ring sideroblasts are classic peripheral blood/marrow findings
- del(5q) syndrome responds strikingly to lenalidomide; SF3B1 mutations predict ring sideroblasts and favorable prognosis; TP53 mutations predict very poor outcomes
- Azacitidine is the standard first-line disease-modifying agent for high-risk MDS ineligible for transplant
- Allo-HSCT is the only cure; few patients are candidates
- Most patients die from complications of pancytopenia (infection, bleeding), not AML transformation
- Transformation to AML occurs in 10-40% of cases
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 107
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 13
- Robbins & Kumar Basic Pathology, Chapter 10
- Goldman-Cecil Medicine, Chapters 167 & 168
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Chapters 9 & 33
Polyglandular autoimmune deficiency syndromes
"polyglandular autoimmune syndrome"[MeSH Terms]
"autoimmune polyglandular syndrome" OR "APS-1" OR "APECED"
Polyglandular Autoimmune (PGA) Deficiency Syndromes
Overview and Classification
| Feature | APS Type 1 | APS Type 2 | APS Type 3 | APS Type 4 |
|---|---|---|---|---|
| Also known as | APECED | Schmidt syndrome | Multiple autoimmune syndrome type 3 | - |
| Inheritance | Autosomal recessive | Polygenic (HLA-linked) | Polygenic | Polygenic |
| Gene | AIRE (chr 21q22) | HLA DR3/DR4, CTLA4, PTPN22, MICA | HLA-linked | HLA-linked |
| Age of onset | Infancy/early childhood | 4th decade (adult) | Adulthood | Adulthood |
| Sex | Equal M:F | F >> M | F >> M | Variable |
| Frequency | Rare (<500 cases reported) | Most common type | Common | Rare |
APS Type 1 (APECED)
Genetics and Pathogenesis
-
Caused by inactivating mutations in the AIRE gene (Autoimmune REgulator), chromosome 21q22 - over 100 mutations described
-
AIRE is expressed in thymic medullary epithelial cells (mTECs), where it drives expression of tissue-specific self-antigens (e.g., insulin) to enable central T-cell tolerance (negative selection of autoreactive T cells)
-
Without AIRE, autoreactive T cells escape to the periphery and destroy target organs
-
Autoantibodies to type I interferons (IFN-α, IFN-ω) are a diagnostic hallmark - present in nearly 100% of cases regardless of AIRE mutation type, and not found in other autoimmune disorders
-
Autoantibodies to IL-17 and IL-22 explain the susceptibility to mucocutaneous candidiasis, as these Th17 cytokines are critical for antifungal defense
-
Higher frequency in Sardinians, Finns, Iranian Jews, Norwegians, and Irish (founder mutations)
-
A non-classical autosomal dominant form (PHD1 domain mutations) has been described with later onset and incomplete expression
-
Harrison's 22E, p. 3132; Robbins & Kumar Pathologic Basis of Disease, p. 2703
Classic Triad (diagnostic criteria: any 2 of 3)
- Chronic mucocutaneous candidiasis (CMC) - virtually always first, affects mouth and nails > skin and esophagus
- Hypoparathyroidism (>85% of cases) - typically second manifestation
- Addison's disease (autoimmune adrenal insufficiency, ~80%) - typically third
Full Clinical Manifestations of APS-1
- Hypoparathyroidism: >85%
- Addison's disease: ~80%
- Gonadal failure (premature ovarian failure): 70% of women (secondary amenorrhea), 25% of men
- Dental enamel hypoplasia: 77%
- Type 1 diabetes mellitus: 23%
- Autoimmune thyroid disease: 18%
-
Alopecia: 40%
-
Pernicious anemia: 31%
-
Vitiligo: 26%
-
Intestinal malabsorption/steatorrhea: ~18-20% (caused by autoantibody-mediated destruction of enteroendocrine cells producing CCK; anti-tryptophan hydroxylase antibodies against enterochromaffin cells)
-
Chronic active hepatitis: 17%
-
Nail dystrophy
-
Ectodermal dysplasia: enamel hypoplasia, nail dystrophy, tympanic membrane calcification
-
Ocular complications: keratitis, blepharitis, corneal opacities, retinitis, ptosis
-
Cerebellar ataxia
-
Asplenism (Howell-Jolly bodies on peripheral blood smear) - unique to APS-1
-
Autoimmune pneumonitis (up to 40% - underrecognized)
-
Obstructive respiratory disease
-
Vitamin B12 malabsorption (autoimmune gastritis)
-
IgA deficiency
-
Goldman-Cecil Medicine, p. 2502; Harrison's 22E, p. 3133
APS Type 2 (Schmidt Syndrome)
Genetics and Pathogenesis
- Polygenic disorder - most common genetic locus: HLA B8, DR3, DR4
- Additional susceptibility genes: CTLA-4 (cytotoxic T-lymphocyte antigen-4), MICA allele 5.1, STAT4, GATA3, PTPN22 (encodes lymphoid tyrosine phosphatase - a variant is enriched in families with T1D + autoimmune thyroid disease)
- Genome-wide association studies (GWAS) have identified numerous additional susceptibility loci
- HLA associations do not predict disease absolutely, even in identical twins - environmental and other genetic factors also influence expression
Classic Components
- Addison's disease (autoimmune adrenal insufficiency) - the anchor diagnosis
- Autoimmune thyroid disease (Hashimoto's thyroiditis or Graves' disease)
- Type 1 diabetes mellitus
- Addison's disease is present in APS-2 but NOT in APS-3
- Mucocutaneous candidiasis, ectodermal dysplasia, and hypoparathyroidism do NOT occur in APS-2
Additional Associations
-
Primary hypogonadism
-
Hypophysitis (autoimmune)
-
Pernicious anemia
-
Vitiligo
-
Celiac disease / dermatitis herpetiformis
-
Alopecia
-
Myasthenia gravis (unique to APS-2; not seen in APS-1)
-
IgA deficiency
-
Idiopathic thrombocytopenia
-
Parkinson's disease
-
Goldman-Cecil Medicine, p. 2502-2503; Harrison's 22E, p. 3133
APS Type 3
- Autoimmune thyroid disease (Hashimoto's thyroiditis or Graves') + another autoimmune condition
- BUT without Addison's disease (the key distinction from APS-2)
- Also called "Multiple Autoimmune Syndrome type 3"
- Associated non-thyroid autoimmune disorders: T1D, pernicious anemia, vitiligo, alopecia, celiac disease, myasthenia gravis, chronic active hepatitis, IgA deficiency
- An expanding category - additional autoimmune manifestations involving GI tract, pancreas, skin, heart, hepatobiliary system, nervous system, and hematologic system
APS Type 4
- Combination of T1D, pernicious anemia, alopecia, vitiligo, or neuromuscular junction disorder
- Without Addison's disease, thyroid disease, or hypoparathyroidism
- Substantial proportion have anti-GAD (glutamic acid decarboxylase) antibodies
- Rare variant
Comparison: APS-1 vs. APS-2
| Feature | APS-1 | APS-2 |
|---|---|---|
| Mucocutaneous candidiasis | Very common (virtually all) | NOT seen |
| Hypoparathyroidism | Common (>85%) | Rare |
| Addison's disease | Common (~80%) | Common |
| Primary hypogonadism | Common (70% women, 25% men) | Occurs |
| Autoimmune thyroid disease | Rare (18%) | Very common |
| Type 1 diabetes | Occurs (23%) | Common |
| Alopecia | Common (40%) | Occurs |
| Keratopathy | Common | Not seen |
| Tympanic membrane calcification | Common | Not seen |
| Autoimmune hepatitis | Occurs | Not seen |
| Autoimmune pneumonitis | Occurs | Not seen |
| Myasthenia gravis | Not seen | Occurs |
| Asplenism | Present (Howell-Jolly bodies) | Not defined |
| Anti-IFN-α/ω antibodies | Yes (nearly 100%) | No |
| Anti-IL-17/IL-22 antibodies | Yes | No |
Other Related Syndromes
| Syndrome | Key Features |
|---|---|
| IPEX | Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked; FOXP3 mutations; regulatory T-cell defect |
| POEMS syndrome | Polyneuropathy, Organomegaly, Endocrinopathy, M-protein, Skin changes |
| DIDMOAD (Wolfram) | Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, Deafness |
| Insulin autoimmune syndrome (Hirata's) | Anti-insulin autoantibodies causing hypoglycemia |
| Thymic tumors | Thymoma associated with various autoimmune endocrinopathies |
Investigations
Diagnostic Autoantibodies
| Condition | Key Autoantibody |
|---|---|
| APS-1 (diagnostic) | Anti-IFN-α, Anti-IFN-ω (nearly 100% sensitive; unique to APS-1) |
| APS-1 candidiasis | Anti-IL-17A, Anti-IL-17F, Anti-IL-22 |
| Addison's disease | Anti-21-hydroxylase, anti-17-hydroxylase |
| Hypoparathyroidism | Anti-CaSR (calcium-sensing receptor); anti-NALP5 |
| Autoimmune thyroid | Anti-TPO (thyroperoxidase), anti-thyroglobulin, anti-TSH-receptor (Graves') |
| Type 1 diabetes | Anti-GAD65, anti-insulin (IAA), anti-IA-2, anti-ZnT8, islet cell antibodies (ICA) |
| Pernicious anemia | Anti-intrinsic factor, anti-parietal cell |
| APS-1 malabsorption | Anti-tryptophan hydroxylase (enterochromaffin cells) |
Clinical Investigations
- APS-1 specific: AIRE gene mutation analysis + anti-IFN-α/ω antibodies (near 100% sensitivity)
- Endocrine workup:
- Morning cortisol + ACTH stimulation test (Addison's)
- Serum calcium, PTH (hypoparathyroidism)
- TSH, free T4, T3 (thyroid disease)
- Fasting glucose, HbA1c, C-peptide (T1D)
- LH, FSH, oestradiol/testosterone (gonadal failure)
- Monitoring for complications:
- FBC (pernicious anemia, asplenism)
- Vitamin B12
- Fecal fat / anti-tissue transglutaminase (malabsorption)
- LFTs + anti-smooth muscle / LKM antibodies (autoimmune hepatitis)
- Echocardiogram (cardiac involvement in POEMS)
Key Diagnostic Pitfalls
- Thyroid hormone replacement in a patient with undiagnosed Addison's disease can precipitate an adrenal crisis by accelerating cortisol metabolism - always screen for adrenal insufficiency before starting thyroxine in hypothyroid patients with autoimmune disease
- Combinations of hypothyroidism + adrenal insufficiency + hypogonadism can mimic hypopituitarism - hormonal testing easily distinguishes
- Components appear asynchronously over years - periodic re-screening is mandatory
- APS-1 siblings should be screened even if only one component disorder is detected
Management
APS-1
- Topical ketoconazole (1-2 weeks) for mild disease
- Oral itraconazole solution for refractory or esophageal disease
- Long-term antifungal prophylaxis often required (chronic oral candidiasis risks leukoplakia and squamous carcinoma)
- Oral elemental calcium: 500-1000 mg every 6-12 hours
- Active vitamin D: calcitriol (1,25-dihydroxyvitamin D) 0.25-2 mcg/day
- Goal: serum calcium in low-normal range (8.0-8.5 mg/dL) to minimize renal stone risk
- Synthetic PTH (1-84) 50-100 mcg SC daily: reduces required doses of calcium and vitamin D
- Intestinal malabsorption complicates management
- Glucocorticoid replacement: hydrocortisone 15-25 mg/day (typically 10mg on waking + 5mg early afternoon) OR cortisone acetate
- Mineralocorticoid replacement: fludrocortisone 0.05-0.2 mg/day
- Sick-day rules: double/triple dose during illness; parenteral hydrocortisone for vomiting/surgery
- HRT (women): combined oestrogen/progesterone until age of natural menopause
- Testosterone replacement (men)
- JAK inhibitors: IFN-γ plays a role in APS-1 organ destruction; ruxolitinib (JAK1/2 inhibitor) has shown early promise in clinical studies for APS-1 (NEJM 2024, [PMID 38810185])
- Targeting the IFN-γ pathway may alter the autoimmune disease course
APS-2, 3, and 4
- Treatment of each component is identical to the sporadic disease in isolation
- No disease-modifying therapy targeting the underlying polyglandular autoimmunity exists
- Regular screening for other components is the cornerstone of management
Screening and Surveillance Recommendations
- Annual: fasting glucose, calcium, cortisol/ACTH stimulation, thyroid function, LFTs, FBC, B12
- Antibody surveillance: anti-21-hydroxylase, anti-thyroid, anti-GAD, anti-gonadal antibodies
- Periodic ophthalmologic assessment (corneal complications)
- Upper GI endoscopy if dysphagia (esophageal candidiasis or stricture)
- Every patient with autoimmune hypothyroidism should have careful assessment for adrenal insufficiency (family history, symptoms, low-threshold testing with short Synacthen test)
- Annual screening: fasting glucose, morning cortisol, thyroid function, anti-21-hydroxylase, anti-GAD, anti-TPO antibodies
- Family members: HLA typing and antibody screening in first-degree relatives
Prognosis
- APS-1: Prognosis of each individual component disorder is similar to its sporadic counterpart; early detection via surveillance improves outcomes. Long-term risks include oral carcinoma (from chronic candidiasis) and complications of poorly treated Addison's disease
- APS-2/3/4: Prognosis of individual components is the same as for their sporadic versions; however, cumulative burden from multiple endocrine deficiencies can reduce quality of life significantly
- Therapy-related APS (from immune checkpoint inhibitors used in cancer therapy) increasingly recognized - similar components but in a specific clinical context
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 401
- Goldman-Cecil Medicine, Chapter 212
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 11
- Frameworks for Internal Medicine, Chapter on Adrenal Insufficiency
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease, Chapter 104
Biological warfare- bioterrorism
bioterrorism biological warfare agents clinical management
bioterrorism Category A B C agents clinical features treatment CDC 2024
Biological Warfare and Bioterrorism
Definition
Historical Context
- Plague was weaponized by the Japanese before World War II (infected fleas released in Chinese cities)
- The former Soviet Union maintained large biological weapons arsenals (including plague, smallpox, anthrax)
- The 2001 anthrax letter attacks in the United States (22 cases, 5 deaths) demonstrated the potential for mail-based delivery
- Unaccounted-for vials of variola (smallpox) found in US laboratories raise ongoing concerns
- From 1981-2018, at least 37 bioterrorist attacks occurred worldwide
CDC Classification of Bioterrorism Agents
Category A - Highest Priority
- Can be easily disseminated or transmitted from person to person
- Cause high mortality
- Have potential to cause public panic and social disruption
- Require special public health preparedness actions
- Can be formulated as stable aerosols for efficient spread
- Anthrax (Bacillus anthracis)
- Smallpox (variola virus)
- Plague (Yersinia pestis)
- Tularemia (Francisella tularensis)
- Botulism (botulinum toxin, Clostridium botulinum)
- Viral hemorrhagic fevers (Ebola, Marburg, Lassa, Machupo, etc.)
Category B - Second Highest Priority
- Moderately easy to disseminate
- Cause moderate morbidity and low mortality
- Require specific diagnostic capacity and surveillance
- Examples: Brucellosis, Q fever, glanders, melioidosis, Vibrio cholerae, ricin toxin, Salmonella, E. coli O157:H7, Staphylococcal enterotoxin B, Venezuelan equine encephalitis
Category C - Emerging Threats
- Emerging pathogens that could be engineered for mass dissemination
- Available, easily produced, high potential for morbidity and mortality
- Examples: Nipah virus, hantavirus, novel influenza strains, Zika virus
Epidemiologic Clues to a Bioterrorism Attack
| Category | Clue |
|---|---|
| Case clustering | Large outbreak in a discrete population; multiple simultaneous or serial epidemics |
| Unusual disease pattern | Disease outside its geographic area or transmission season; vector-borne disease where vector is absent |
| Atypical presentation | Disease by unusual route (e.g., inhalational anthrax, pneumonic plague); more severe than expected |
| Treatment failure | Failure to respond to standard therapy; unusual antibiotic resistance pattern |
| Rare disease | Single case of smallpox, inhalational anthrax, or viral hemorrhagic fever |
| Spatial pattern | Noncontiguous outbreaks simultaneously; downwind pattern of cases |
| Laboratory clues | Unusual organism strains; same genetic type from different sources/times |
| Human-animal discordance | Zoonotic disease in humans but not animals |
| Direct evidence | Suspicious spray devices, tampered letters/packages, intelligence of release |
Category A Agents: Detailed Clinical Profiles
1. ANTHRAX (Bacillus anthracis)
Three Clinical Forms:
- Painless pruritic papule → vesicle → central black eschar (anthrakis = coal in Greek) with surrounding disproportionate edema
- Case-fatality rate: 20% if untreated
- Incubation: average 1-6 days (can be up to 43 days)
- Prodrome (1-3 days): fever, profound drenching sweats, nausea, vomiting, diarrhea, cough, chest pain
- Rapid deterioration: increasing dyspnea, stridor, cyanosis, respiratory failure
- Hallmark: widened mediastinum on CXR (~60% of cases) from hemorrhagic mediastinitis
- Large hemorrhagic pleural effusions and infiltrates
- Bacteremic seeding of GI tract and meninges
- Untreated case-fatality rate: approaches 100%
- Fever, abdominal pain, diarrhea, hematochezia, hematemesis, ascites
- Paracentesis: hemorrhagic ascites
- Case-fatality rate: 50% or more
- Ciprofloxacin 400 mg q8h IV (or levofloxacin 750 mg q24h, or moxifloxacin 400 mg q24h)
- + Meropenem 2 g q8h IV (or imipenem 1 g q6h, or doripenem 500 mg q8h)
- + Linezolid 600 mg q12h (or clindamycin 900 mg q8h, or rifampin 600 mg q12h)
- Fluoroquinolone or carbapenem or vancomycin + clindamycin or linezolid or doxycycline
- Raxibacumab (monoclonal antibody, 40 mg/kg IV) - also licensed for prophylaxis
- Obiltoxaximab - second licensed monoclonal antibody
- Anthrax antitoxin (equine heptavalent)
- Pleural fluid drainage (improves survival)
2. SMALLPOX (Variola virus)
- Incubation: 7-17 days (usually 12-14)
- Prodrome (2-3 days): sudden onset fever (38-40°C), headache, backache (prominent), vomiting, prostration
- Rash (centrifugal - face and extremities > trunk): synchronous progression in ALL lesions simultaneously:
- Macules → papules (day 1-2) → vesicles (day 4-5) → umbilicated pustules (day 7) → scabs (day 14)
- Lesions are deep-seated, painful
- All in same stage of development at any time point
- Contagious from onset of rash until all scabs have separated
- Hemorrhagic smallpox / flat-type smallpox: case-fatality rate approaches 100%
- Variola minor: mild variant, CFR ~1%
- Variola major (typical): CFR ~30%
| Feature | Smallpox | Chickenpox |
|---|---|---|
| Distribution | Centrifugal (face + extremities > trunk) | Centripetal (trunk > extremities) |
| Lesion progression | Synchronous (all same stage) | Asynchronous (crops at different stages) |
| Lesion depth | Deep-seated, painful | Superficial |
| Pre-rash contagiousness | No | Yes |
| Backache | Prominent | Mild |
- Primary: supportive care
- Tecovirimat (600 mg orally q12h × 14 days) - FDA approved for all poxvirus infections; in strategic national stockpile
- Brincidofovir (4 mg/kg once weekly × 2 doses, max 200 mg) - oral cidofovir analog; FDA approved
- Vaccinia virus-based vaccine (ACAM2000) stockpiled for entire US population
- Post-exposure vaccination within 4 days can prevent or ameliorate disease
- Ring vaccination (surveillance + containment strategy used during eradication)
- Contraindications: immunocompromised, eczema, significant exfoliative skin disease
3. PLAGUE (Yersinia pestis)
- Swollen, extremely tender lymph nodes (bubo) - most commonly inguinal/femoral
- Fever, headache, malaise
- Can progress to septicemic plague
- Necrosis of nose, ears, digits (temperature-dependent coagulase = "Black Death" appearance)
- Untreated CFR: ~50%
- Clusters of multiple patients with hemoptysis (purulent → hemorrhagic sputum)
- Human-to-human transmission by respiratory droplets
- Untreated CFR: nearly 100%
- Streptomycin 1 g IM BD × 7-10 days (preferred if available)
- Gentamicin 5 mg/kg/day IV/IM
- Ciprofloxacin 400 mg q12h IV, or doxycycline 200 mg then 100 mg q12h, or levofloxacin 500 mg q24h
4. TULAREMIA (Francisella tularensis)
- Incubation: 2-10 days
- Six clinical forms; most relevant after aerosol release: pneumonic and typhoidal forms (analogous to plague)
- Fever, severe exhaustion, substernal chest pain, nonproductive cough, weight loss
- Glandular form resembles bubonic plague
- Streptomycin or gentamicin × 7-10 days (drugs of choice)
- Ciprofloxacin (newer alternative)
- Doxycycline/tetracycline - second-line (higher relapse rate)
- CFR: 30% untreated; <1% with treatment
5. BOTULISM (Botulinum toxin, Clostridium botulinum)
- Latent period: hours to several days
- Cranial nerve involvement first: ptosis, blurred vision, diplopia, photophobia, dry mouth
- Descending symmetrical flaccid paralysis (cranial → neck → arms → respiratory muscles → legs)
- Dysphonia, dysphagia
- Death from respiratory muscle failure
- Not contagious (standard precautions only)
- Supportive care (ventilatory support - may last months)
- Heptavalent botulinum antitoxin (HBAT, serotypes A-G): halts but does NOT reverse paralysis - give as soon as possible
- No antimicrobials needed (toxin-mediated, not infectious)
6. VIRAL HEMORRHAGIC FEVERS (VHF)
| Family | Viruses | Key Features |
|---|---|---|
| Filoviruses | Ebola (CFR 18-90%), Marburg (23-70%) | Greatest bioterrorism concern; aerosol infectious; nosocomial transmission |
| Arenaviruses | Lassa (CFR 1-2%), Junin (30%), Machupo (25-35%) | Ribavirin effective; immune plasma |
| Bunyaviruses | Crimean-Congo HF (30%), Rift Valley fever, Hantaviruses | CCHF has nosocomial risk |
| Flaviviruses | Yellow fever (3-12%+), Dengue | Yellow fever vaccine available |
- Febrile illness with malaise, myalgias, headache, vomiting, diarrhea
- Hemorrhagic features (massive hemorrhage in <50%): purpura, petechiae, mucosal bleeding
- Hypotension, shock
- Multiorgan failure
| Agent | Treatment |
|---|---|
| Ebola (Zaire) | REGN-EB3 (atoltivimab/maftivimab/odesivimab) or mAb114 (ansuvimab) - two FDA-approved monoclonal antibody combinations; Ervebo vaccine |
| Lassa, New World arenaviruses, CCHF, HFRS | Ribavirin 30 mg/kg load → 16 mg/kg q6h × 4 days → 8 mg/kg q8h × 6 days |
| Other VHF | Supportive care; no proven specific therapy |
Category B and C Agents: Summary
Key Category B Agents
| Agent | Disease | Treatment |
|---|---|---|
| Brucella spp. | Brucellosis (undulant fever) | Doxycycline + rifampin/streptomycin |
| Coxiella burnetii | Q fever | Doxycycline |
| Burkholderia mallei | Glanders | Ceftazidime or imipenem |
| Burkholderia pseudomallei | Melioidosis | Ceftazidime or meropenem |
| Ricin toxin | Cell death, respiratory failure | Supportive (no antidote) |
| Staphylococcal enterotoxin B | Toxic shock/food poisoning | Supportive |
| Clostridium perfringens epsilon toxin | Pulmonary edema, necrosis | Supportive |
| T-2 mycotoxin | Skin/mucous membrane necrosis | Supportive |
Category C Examples
- Nipah virus (>75% CFR in some outbreaks)
- Hantavirus (Sin Nombre)
- Novel respiratory pathogens (engineered influenza)
- Zika virus
General Principles of Response
Immediate Clinical Response
- Recognize unusual presentations or disease clustering
- Isolate the patient with appropriate precautions (see individual agent above)
- Notify immediately:
- Hospital infection control
- Local/state public health department
- CDC Emergency Operations Center: 770-488-7100
- FBI (if criminal act suspected)
- Collect specimens before starting treatment if possible
- Treat empirically while awaiting results - delays are fatal
Post-Exposure Prophylaxis Overview
| Agent | Prophylaxis |
|---|---|
| Anthrax | Ciprofloxacin 500 mg BD oral OR doxycycline 100 mg BD oral × 60 days ± vaccine |
| Smallpox | Vaccination within 4 days of exposure; tecovirimat if vaccine unavailable |
| Plague | Ciprofloxacin 500-750 mg BD oral OR doxycycline 100 mg BD × 7 days |
| Tularemia | Ciprofloxacin or doxycycline × 14 days |
Decontamination
- Biological agents are generally removed by soap and water
- Remove and bag clothing
- Universal precautions during decontamination
- Pediatric decontamination with family unit where possible
Smallpox vs. Chickenpox: Key Differentiating Points
| Feature | Smallpox (Variola) | Chickenpox (Varicella) |
|---|---|---|
| Rash distribution | Centrifugal (face, extremities) | Centripetal (trunk) |
| Lesion stages | All synchronous | Multiple stages simultaneously |
| Lesion depth | Deep-seated, painful | Superficial |
| Backache | Prominent | Mild |
| Pre-rash transmission | No | Yes |
| Scab = end of contagion | Yes | Yes |
| Palms/soles involved | Yes | Rare |
Principles of Preparedness
- Maintain high clinical suspicion for unusual disease presentations
- Know your local reporting chain - physicians are first responders
- Laboratory notification is critical for dangerous organisms (F. tularensis, Y. pestis)
- Strategic national stockpile (SNS) contains antibiotics, antitoxins, vaccines for mass casualty response
- Ring vaccination remains the containment strategy of choice for smallpox
- Infection control measures are agent-specific and must be applied rapidly
- Goldman-Cecil Medicine, Chapter 19 (Kortepeter & Cieslak) - Primary source
- Parikh's Textbook of Medical Jurisprudence, Forensic Medicine and Toxicology, Chapter 47
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 8
- Jawetz, Melnick & Adelberg's Medical Microbiology, Chapter on Bioterrorism Agents
- CDC Bioterrorism Diseases/Agents List
- USAMRIID's Medical Management of Biological Casualties Handbook, 9th ed., 2020
Opioid related disorders
"opioid use disorder"[MeSH Terms] AND treatment
"opioid use disorder" buprenorphine methadone treatment
Opioid-Related Disorders
Classification (DSM-5)
- Opioid Use Disorder (OUD)
- Opioid Intoxication
- Opioid Withdrawal
- Opioid-Induced Disorders (intoxication delirium, psychotic disorder, mood disorder, sleep disorder, sexual dysfunction)
- Unspecified Opioid-Related Disorder
Epidemiology and the Opioid Crisis
- 1999-2010: Increased prescription opioids (peak oxycodone/hydrocodone sales in 2012); aggressive pharmaceutical marketing; overprescribing of extended-release oxycodone
- From 2010: Rise in heroin overdose deaths (as prescription opioids became restricted)
- From 2013: Surge in synthetic opioids - particularly fentanyl (50-100× more potent than morphine, 10× more potent than heroin) and fentanyl analogues, now the leading driver of overdose deaths
Neuropharmacology
- Pain pathways: strong analgesia
- Reward pathway: euphoria and liability for misuse
- Respiratory center: inhibition - risk of lethal overdose
- Locus coeruleus: inhibition of noradrenergic tone (withdrawal symptoms result from rebound hyperactivity)
1. Opioid Use Disorder (OUD)
DSM-5 Diagnostic Criteria
| Domain | Criterion |
|---|---|
| Impaired control | 1. Taken in larger amounts or over longer period than intended |
| 2. Persistent desire or unsuccessful efforts to cut down or control use | |
| 3. Great deal of time spent obtaining, using, or recovering from opioids | |
| 4. Craving - strong desire or urge to use | |
| Social impairment | 5. Failure to fulfill major role obligations (work, school, home) |
| 6. Continued use despite persistent social/interpersonal problems | |
| 7. Important social, occupational, or recreational activities given up | |
| Risky use | 8. Recurrent use in physically hazardous situations |
| 9. Continued use despite physical or psychological problems | |
| Pharmacological | 10. Tolerance (need for markedly increased amounts; diminished effect with same amount) |
| 11. Withdrawal (characteristic syndrome; opioids taken to relieve or avoid withdrawal) |
- Mild: 2-3 criteria
- Moderate: 4-5 criteria
- High: ≥6 criteria
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 4258
Specifiers
- In early remission: 3-12 months without criteria (except craving)
- In sustained remission: ≥12 months without criteria
- On maintenance therapy (methadone/buprenorphine)
- In a controlled environment
2. Opioid Intoxication
- Recent opioid use
- Classic triad:
- Miosis (pinpoint pupils) - bilateral, symmetric
- CNS depression - drowsiness, stupor, coma ("on the nod")
- Respiratory depression - reduced rate and depth, apnea
- Additional features: slurred speech, psychomotor retardation, impaired attention and memory, bradycardia, hypotension, hypothermia, decreased bowel sounds, urinary retention
- Maladaptive behavioral changes: euphoria followed by apathy, dysphoria, psychomotor retardation
3. Opioid Withdrawal
Mechanism
Withdrawal Symptoms and Signs
| Symptoms (subjective) | Signs (objective) |
|---|---|
| Craving for opioids | Mydriasis (pupillary dilation) |
| Restlessness, irritability, anxiety | Sweating, diaphoresis |
| Increased sensitivity to pain (hyperalgesia) | Piloerection ("gooseflesh" - cold turkey) |
| Nausea, abdominal cramps | Tachycardia |
| Muscle aches, bone pain | Vomiting, diarrhea |
| Dysphoric mood, depression | Hypertension |
| Insomnia | Yawning |
| Weakness, tremor | Fever, temperature dysregulation |
| Rhinorrhea, lacrimation, sneezing |
Onset and Duration by Opioid
| Opioid | Withdrawal Onset | Peak Intensity | Duration |
|---|---|---|---|
| Heroin | 6-8 hours after last dose | Day 2-3 | 7-10 days |
| Morphine | 6-8 hours | Day 2-3 | 7-10 days |
| Methadone | 1-3 days | Day 3-5 | 10-14 days |
| Buprenorphine | 2-4 days | Day 3-5 | Variable |
| Fentanyl | 2-4 hours | Day 1-2 | ~4-5 days |
Opioid Overdose - Clinical Emergency
Classic Triad (Toxidrome)
- Coma / CNS depression (unresponsive, stupor)
- Miosis (pinpoint pupils, bilateral)
- Respiratory depression (rate <12/min, shallow, apnea)
Immediate Management
-
Airway - open airway, clear secretions; insert oral/nasal airway; intubate if needed
-
Ventilation - bag-mask ventilation until naloxone takes effect
-
Naloxone (Narcan) - competitive MOR antagonist:
- IV: 0.4-2 mg IV, titrate to adequate respiration (not full reversal - prevents precipitated withdrawal)
- Initial dose: ~0.8 mg per 70 kg body weight; repeat every few minutes
- Nasal spray: 4 mg intranasal (2 mg/mL has similar efficacy to 2 mg IM)
- Signs of response: increased respiratory rate + pupillary dilation
- Duration of naloxone (1-2 hours) is shorter than most opioids - re-dosing or infusion required
- If no response after 4-5 mg: consider non-opioid causes of CNS depression
- Caution: In opioid-dependent patients, excessive naloxone precipitates severe withdrawal
-
Monitor - continue monitoring for re-narcotization (especially with long-acting opioids like methadone, fentanyl)
-
IV access - dextrose if hypoglycaemia possible; thiamine
-
Fentanyl overdose: may require multiple or higher doses of naloxone due to potency and kinetics
-
History of overdose
-
History of substance use disorder
-
Opioid dose ≥50 morphine milligram equivalents (MME)/day
-
Concurrent benzodiazepine use
-
Recently released from incarceration or completed detoxification (lost tolerance)
-
Miller's Anesthesia 10e, p. 2884; Kaplan & Sadock's Synopsis, p. 930
Laboratory and Investigations
Urine Drug Testing (UDT)
| Substance | Detection Window in Urine |
|---|---|
| Heroin (6-acetylmorphine - specific metabolite) | Up to 8 hours after use |
| Heroin (detected as morphine) | 1-3 days |
| Morphine, codeine, hydrocodone | 1-3 days |
| Methadone | Up to 4 days (single dose); longer with repeated use |
| Fentanyl, buprenorphine, tramadol, oxycodone | Require specific immunoassay - not detected on standard opioid screen |
| Poppy seeds | May produce false positive for morphine/codeine |
-
Drug metabolites first detectable 3-6 hours after use
-
Saliva testing: similar sensitivity to urine; potentially shorter detection window
-
Hair testing: detects longer-term use (months)
-
No biomarker currently exists to diagnose OUD
-
Kaplan & Sadock's Comprehensive Textbook, p. 4258
Management of Opioid Use Disorder
Treatment Goals (not uniform - individualized)
- Protection against overdose and death
- Cessation of illicit opioid use
- Elimination of craving and withdrawal
- Improvement in physical and psychological health
- Eventual abstinence (long-term recovery goal)
- Harm reduction (immediate realistic goal)
A. Medications for OUD (MOUD) / Medication-Assisted Treatment (MAT)
1. METHADONE (Full MOR Agonist)
- Starting dose: 20-30 mg oral (to avoid oversedation)
- Increase by 10 mg/day based on response
- Optimal maintenance dose: 60-120 mg/day (doses >60 mg show significantly improved outcomes)
- Available as: oral solution (primary), tablets, injectable
- QT prolongation - screen ECG before starting and monitor (particularly at doses >100 mg/day or with other QT-prolonging drugs)
- Constipation, excessive sweating
- Decreased libido and sexual dysfunction (reduces testosterone and FSH)
- Sleep abnormalities (insomnia, nightmares - often during first months)
- Sedation, respiratory depression (risk with rapid dose escalation)
- Drug interactions (CYP3A4): rifampin, phenytoin, carbamazepine, darunavir, efavirenz → decrease methadone levels → withdrawal; fluconazole/ketoconazole → increase levels → toxicity
2. BUPRENORPHINE (Partial MOR Agonist)
- Partial agonist at MOR - produces agonist effects at lower doses, then ceiling effect on respiratory depression (→ safer margin than methadone)
- Very high affinity for MOR - displaces other opioids; blocks effects of heroin
- If given while patient is opioid-dependent, displaces current agonist → precipitated withdrawal
- Low dissociation from receptor → long duration of action
| Formulation | Brand | Notes |
|---|---|---|
| Buprenorphine/naloxone sublingual (4:1 ratio) | Suboxone, Zubsolv | Preferred - naloxone prevents diversion and IV misuse |
| Buprenorphine sublingual | Subutex | Reserved for pregnancy, directly observed therapy |
| Extended-release SC injection | Sublocade | 300 mg q4weeks × 2, then 100 mg q4weeks |
| Buprenorphine/naloxone film | Generic | Sublingual or buccal |
- Naloxone has low oral bioavailability (1-3%) - when taken sublingual, it has minimal systemic effect
- If tampered with and injected, naloxone becomes bioavailable → precipitates withdrawal in opioid-dependent users → deters diversion
- Induction: Patients must be in mild-moderate withdrawal (COWS score ≥8-12) before first dose to avoid precipitating withdrawal
- Starting dose: 2-4 mg sublingual; titrate up by 2 mg every 1-2 hours as tolerated to 8 mg on day 1
- Maintenance: typically 12-24 mg/day; max 24 mg/day (sublingual)
3. EXTENDED-RELEASE NALTREXONE (Full MOR Antagonist)
- Oral naltrexone: 50 mg daily or 100/100/150 mg Mon/Wed/Fri (poor adherence)
- Extended-release IM naltrexone (Vivitrol): 380 mg IM every 4 weeks; more than double the success rate of oral naltrexone
- Patient must be fully opioid-free for 7-10 days (short-acting opioids) or longer (methadone 7-14 days minimum)
- Urine drug screen must be negative
- Naloxone challenge test (0.8-1.2 mg IM naloxone) to confirm no residual physical dependence before initiating full dose
Summary Table: MOUD Comparison
| Methadone | Buprenorphine/naloxone | XR-Naltrexone | |
|---|---|---|---|
| Mechanism | Full agonist | Partial agonist | Full antagonist |
| Route | Oral | Sublingual | IM injection |
| Starting dose | 20-30 mg/day | 4-8 mg/day | 380 mg q4weeks |
| Setting | OTP clinic only | Office-based | Office-based |
| Overdose risk | Higher | Lower (ceiling effect) | None |
| Diversion risk | High | Moderate | None |
| Withdrawal before initiation | Not required | Required (COWS ≥8) | Required (7-10 days) |
| Key side effect | QT prolongation | Precipitated withdrawal | Opioid withdrawal if abstinence incomplete |
| Evidence strength | Strongest | Strong | Strong |
B. Management of Acute Opioid Withdrawal (Detoxification)
- Transfer to methadone 20-30 mg oral, reduce by 20%/day
- Or buprenorphine 2 mg sublingual (titrate to 8 mg day 1), then gradual taper
- Most common and effective approach
- Clonidine 0.1-0.3 mg orally every hour (up to 4 doses); off-label; reduces autonomic symptoms (rhinorrhea, lacrimation, tachycardia, hypertension) but does NOT relieve aches or craving
- Lofexidine (Lucemyra) 3.2 mg/day in 4 divided doses - FDA-approved specifically for opioid withdrawal; similar to clonidine
- Key side effect: postural hypotension
- Adjuncts for symptom relief: loperamide (diarrhea), NSAIDs/acetaminophen (myalgia), antiemetics (nausea), benzodiazepines (anxiety/insomnia - use cautiously)
- Acupuncture, transcranial magnetic stimulation, auricular vagus nerve stimulation - not yet validated for clinical use
C. Psychosocial Interventions
- Structured, goal-oriented; addresses drug use as learned behavior
- Teaches coping skills and cognitive strategies; evidence for effectiveness in OUD
- Reduces HIV risk behaviors; particularly effective in patients with comorbid depression
- Works synergistically with pharmacotherapy
- Tangible rewards (vouchers, not cash) for positive behaviors (abstinence, treatment attendance, medication adherence)
- Requires monitored urine collection; objective index of progress
- Peer support; spirituality-based; widely available; observational evidence of benefit
- Limitation: emphasizes abstinence from all psychoactive substances → patients on opioid agonists may be ostracized
- Methadone Anonymous (MA): 12-step variant specifically for patients on methadone
- Regular counseling reduces illicit drug use; quality of counselor-patient relationship significantly impacts outcomes
- Improves medication compliance; enhances treatment outcomes
D. Harm Reduction Strategies
- Needle/syringe exchange programs - reduce HIV, hepatitis B and C transmission
- Naloxone co-prescription and distribution to patients and families
- Supervised consumption sites (available in Canada, some European countries)
- Fentanyl test strips - allow users to test drugs for fentanyl contamination
- Treatment of infectious complications: hepatitis C (highly curable with DAA therapy), HIV treatment with ART, endocarditis management, skin abscess care
Comorbidities
- ~70% of men and ~75% of women with lifetime OUD have an additional psychiatric disorder
- Most common: mood disorders (depression, bipolar), antisocial personality disorder, anxiety disorders, alcohol use disorder
- OUD + depression → CBT or supportive psychotherapy superior to drug counseling alone
- Infectious: HIV, hepatitis B and C, tuberculosis, endocarditis (especially tricuspid), septic arthritis, skin abscesses, necrotizing fasciitis
- Pulmonary: aspiration pneumonia, tuberculosis, pulmonary hypertension (talc granulomas)
- Vascular: deep vein thrombosis, thrombophlebitis, pseudoaneurysms
- Neurological: cerebral abscess, meningitis, myelopathy
Opioid-Induced DSM-5 Disorders (Separate Diagnoses)
| Disorder | Notes |
|---|---|
| Opioid intoxication delirium | High doses, combinations with other CNS depressants, or pre-existing CNS disease |
| Opioid-induced psychotic disorder | Hallucinations and/or delusions during intoxication |
| Opioid-induced mood disorder | Manic, depressed, or mixed symptoms; irritability, expansiveness, depression combined |
| Opioid-induced sleep disorder | Hypersomnia or insomnia; common in chronic users |
| Opioid-induced sexual dysfunction | Erectile dysfunction, orgasmic difficulties (opioids reduce testosterone and FSH) |
| Unspecified opioid-related disorder | Significant symptoms not meeting full criteria for other categories |
Special Populations
- Methadone or buprenorphine (without naloxone) are recommended - do NOT abruptly withdraw (risk of fetal distress and preterm labour)
- Neonatal opioid withdrawal syndrome (NOWS/NAS) is expected and manageable - NOT an indication to withhold MOUD
- Naltrexone: insufficient safety data; generally avoided
- Breastfeeding: methadone and buprenorphine compatible with breastfeeding (minimal transfer)
- Period of extremely high overdose risk (lost tolerance to opioids)
- Naloxone rescue kits should be provided; MOUD should be initiated before release
Prognosis and Relapse Prevention
- Detoxification alone has high relapse rates - without MOUD, most patients relapse within days to weeks
- Protracted withdrawal syndrome (anxiety, insomnia, drug craving) persists for up to 6 months after acute withdrawal
- Long-term MOUD reduces mortality (methadone reduces all-cause mortality during treatment; risk increases when treatment is stopped)
- OUD is a chronic relapsing condition - relapses are expected and should not end treatment
- 2026 JAMA review (Harris et al., PMID 41671014) provides updated guidance on medications for OUD, opioid withdrawal, and opioid overdose
- Kaplan & Sadock's Synopsis of Psychiatry, Chapter 4
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, Chapters 11.9 and Treatment of OUD
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 28
- Tintinalli's Emergency Medicine, Chapter 24
- Miller's Anesthesia 10e, Chapter 46
- Adams & Victor's Principles of Neurology 12e
- Harris MTH et al. Medications for OUD, withdrawal, and overdose: A Review. JAMA 2026 Mar. [PMID 41671014]
- Yakovenko I et al. Management of OUD: 2024 update to the national clinical practice guideline. CMAJ 2024 Nov. [PMID 39532476]
- Englander H et al. Caring for hospitalized adults with OUD in the era of fentanyl. JAMA Intern Med 2024. [PMID 38683591]
Oncologic emergencies
"oncologic emergencies" management review
Oncologic Emergencies
Classification
| Category | Examples |
|---|---|
| Structural/Obstructive | Superior vena cava syndrome, Spinal cord compression, Airway obstruction, Cardiac tamponade, Increased intracranial pressure, Neoplastic meningitis, Intestinal obstruction |
| Metabolic | Hypercalcemia, SIADH/Hyponatremia, Tumor lysis syndrome, Lactic acidosis, Hypoglycemia |
| Hematological | Febrile neutropenia, Hyperleukocytosis/Leukostasis, DIC, Massive hemoptysis |
1. Superior Vena Cava Syndrome (SVCS)
Causes
- Malignant (~60%): Lung cancer (particularly SCLC and squamous cell histology) accounts for ~85% of malignant SVCS; lymphoma (Hodgkin's involves mediastinum more commonly but rarely causes SVCS); metastatic disease (testicular, breast)
- Benign (~40%): Intravascular devices (central venous catheters, pacemaker/defibrillator leads), aortic aneurysm, fibrosing mediastinitis, thyromegaly, Behçet's syndrome
- Young man + mediastinal mass = lymphoma vs. primary mediastinal germ cell tumor
Clinical Features
- Facial and neck swelling (especially periorbital, worsened when supine)
- Dyspnea, cough
- Dilated neck veins and collateral chest wall veins
- Cyanosis and edema of face, arms, and chest
- Hoarseness, tongue swelling, nasal congestion, epistaxis, hemoptysis, dysphagia, headache, dizziness, syncope
- Severe: proptosis, glossal/laryngeal edema, obtundation (signs of cerebral edema - poorer prognosis)
- Bending forward or lying down worsens symptoms
- Symptoms are usually progressive but may improve as collateral circulation develops
Investigations
- CXR: Widening of superior mediastinum (right side most common); pleural effusion in 25% (often exudative, right-sided)
- CT chest (contrast): Most reliable - shows mediastinal anatomy, site of obstruction, collateral vessels, identifies tissue biopsy site
- Venography: useful for planning endovascular intervention
- Tissue diagnosis (if not already established): bronchoscopy, CT-guided biopsy, mediastinoscopy, thoracotomy
Treatment
- Airway emergency: head elevation, O2, diuretics, low-salt diet; glucocorticoids (limited role except for lymphoma)
- Endovascular stenting: now first-line for rapid symptom relief; especially for life-threatening symptoms (cerebral edema, laryngeal edema, postural hypotension, coma); complications include hematoma, SVC perforation, stent migration, PE, heart failure post-stenting
- Radiation therapy: primary treatment for NSCLC and solid tumor metastases
- Chemotherapy: for SCLC, lymphoma, germ cell tumors (chemosensitive)
- Anticoagulation ± fibrinolysis: for catheter-related thrombotic SVCS
- SVCS recurs in 10-30%; re-palliation with repeat stenting
- Harrison's 22E, p. 629
2. Malignant Spinal Cord Compression (MSCC)
Causes and Sites
- Most common tumors: Lung, breast, prostate (most frequent); multiple myeloma, lymphoma, melanoma, renal, GU cancers
- Sites: Thoracic spine 70% > Lumbosacral 20% > Cervical 10%
- Mechanism: vertebral body/pedicle metastases → extradural compression of dura; direct paravertebral extension through intervertebral foramen (lymphoma, myeloma); rarely intramedullary hematogenous metastasis
Clinical Features (Progressive)
- Back pain (first symptom in most; present days to months before neurological deficit)
- Worsens with movement, coughing, sneezing
- Worsens when supine (distinguishes from disc disease which typically improves supine)
- Radicular pain in cervical/lumbosacral area (unilateral or bilateral)
- Thoracic radicular pain: tight band-like constriction around thorax and abdomen
- Lhermitte's sign: electric sensation down back and limbs on neck flexion/extension
- Motor weakness, spasticity, extensor plantar response (Babinski)
- Sensory loss (pinprick and vibration); upper level of sensory loss is 1-2 vertebrae below compression
- Autonomic dysfunction (late): bladder/bowel incontinence, decreased anal tone, distended bladder, absent bulbocavernosus reflex - poor prognostic sign
- Ataxia of gait (spinocerebellar involvement)
Investigations
- MRI full spine (gold standard): imaging procedure of choice; multiple metastases in 25%; T1 shows cord/CSF/extradural lesions; T2 shows intramedullary pathology; Gadolinium-enhanced for intramedullary disease
- Plain radiographs: "winking owl" sign (pedicle erosion) earliest sign; insensitive overall
- CT myelography: reserved for patients who cannot undergo MRI
- Bone scan: sensitive but less specific
Treatment (NOMS Framework - Memorial Sloan Kettering)
- Neurologic: degree of epidural compression, myelopathy, radiculopathy
- Oncologic: radiosensitivity of tumor type
- Mechanical: spine stability, retropulsed bone fragments
- Systemic: extent of disease, comorbidities
- Dexamethasone (high-dose corticosteroids): 16-24 mg IV loading dose, then 4 mg q6h - reduces vasogenic edema; start immediately in all symptomatic patients
- Urgent neurosurgery and radiation oncology consultation
- Radiation therapy (RT) + glucocorticoids: initial treatment of choice for most patients with radiosensitive tumors
- Stereotactic body RT (SBRT): preferred for radioresistant tumors (renal, melanoma, sarcoma); requires 2-3mm margin from spinal cord
- Surgery (separation surgery): followed by SBRT for:
- High-grade compression from radioresistant tumors
- Mechanical spinal instability
- Retropulsion of bone fragments
- Rapid neurological deterioration
- Unknown primary requiring histological diagnosis
- Chemotherapy: for chemosensitive tumors (e.g., lymphoma) who have had prior RT
- Vertebroplasty/Kyphoplasty: for painful pathologic compression fractures without spinal instability
- Prognosis: ambulatory status at time of treatment is the strongest predictor of outcome; rapid onset is a poor prognostic feature
- Harrison's 22E, p. 631-633
3. Cardiac Tamponade
Causes
- Malignant pericardial disease found in 5-10% of cancer patients at autopsy
- Most common tumors: lung cancer, breast cancer, leukemia, lymphoma
- ~50% of symptomatic pericardial disease in cancer patients is NOT malignant: radiation pericarditis (acute <months or chronic up to 20 years post-RT), drug-induced (all-trans retinoic acid, arsenic trioxide, imatinib), immune checkpoint inhibitor-related, infection, hypothyroidism, autoimmune, idiopathic
Clinical Features
- Dyspnea, cough, chest pain, orthopnea, weakness
- Beck's triad (classic tamponade): hypotension, JVD, muffled heart sounds
- Pulsus paradoxus (>10 mmHg fall in systolic BP with inspiration)
- Pulsus alternans, friction rub (less common in malignant disease)
- Tachycardia, peripheral edema, hepatomegaly, cyanosis, pleural effusion
Investigations
- Echocardiography: most helpful diagnostic test; shows effusion, diastolic collapse of RV/RA
- CXR: enlarged cardiac silhouette ("water bottle heart"), 90% show abnormalities
- ECG: low voltage, electrical alternans (alternating QRS amplitude)
- Pericardial fluid: serous, serosanguineous, or hemorrhagic; cytology diagnostic in most patients for malignant etiology
- CT chest: identifies concomitant thoracic neoplasm
Treatment
- Life-threatening tamponade: immediate pericardiocentesis (may need echo-guided)
- Recurrence rate after percutaneous drainage ~20%
- Sclerotherapy (bleomycin, mitomycin C, tetracycline instillation): reduces recurrence
- Subxiphoid pericardiotomy (under local anesthesia): for drainage + biopsy
- Thoracoscopic pericardial fenestration (pericardial window): 60% of malignant effusions recur after this procedure
- Radiation/systemic chemotherapy: for underlying malignancy
- Warning: drainage can paradoxically cause worsening hemodynamic instability in ~10% ("low cardiac output syndrome")
- Harrison's 22E, p. 630
4. Increased Intracranial Pressure / Brain Metastases
Common Primary Tumors
- 25% of cancer patients die with intracranial metastases
- Hemorrhagic metastases: melanoma, germ cell tumors, renal cell carcinoma (particularly high risk)
- NSCLC with EGFR/ALK mutations and lobular/triple-negative breast cancer: high risk
Clinical Features
- Headache (often worse in morning), nausea/vomiting, behavioral changes, focal neurological deficits, seizures
- Abrupt onset (mimicking stroke) may indicate hemorrhage into metastasis
- Papilledema, neck stiffness, visual disturbances (with raised ICP)
- Herniation syndromes with mass enlargement: uncal, transtentorial, subfalcine
Investigations
- Gadolinium-enhanced MRI brain: superior to CT; multiple enhancing lesions with surrounding edema; more sensitive for meningeal involvement, small lesions, brainstem/cerebellar lesions
- CT brain if MRI not available urgently
Treatment
- Dexamethasone (first-line for all symptomatic patients): 10-16 mg loading dose IV, then 4 mg q6h; reduces vasogenic edema; caution - may reduce immunotherapy efficacy
- Bevacizumab: for patients unable to wean off steroids or on immunotherapy with symptomatic edema
- Single brain metastasis + controlled extracranial disease: surgical resection + SRS to resection cavity
- 1-4 brain metastases + stable systemic disease: SRS preferred over WBRT (WBRT associated with cognitive dysfunction)
- Multiple metastases: SRS increasingly used; WBRT as alternative; targeted therapy/immunotherapy for appropriate tumor types (EGFR-TKIs for NSCLC, anti-PD1 for melanoma)
- Hydrocephalus: VP shunt placement; ventriculostomy in acute deterioration
- Levetiracetam: seizure management (non-enzyme-inducing; does not alter chemotherapy metabolism)
- Harrison's 22E, p. 633
5. Tumor Lysis Syndrome (TLS)
Definition and Pathophysiology
Cairo-Bishop Criteria
| Parameter | Laboratory TLS Threshold |
|---|---|
| Uric acid | ≥8 mg/dL or 25% increase from baseline |
| Potassium | ≥6.0 mEq/L or 25% increase |
| Phosphorus (adults) | ≥4.5 mg/dL or 25% increase |
| Calcium | ≤7 mg/dL or 25% decrease |
- Creatinine >1.5× upper limit of normal
- Cardiac dysrhythmia or sudden death
- Seizure
Clinical Manifestations
- Hyperkalemia: arrhythmias, cardiac arrest
- Hyperphosphatemia + Hypocalcemia: tetany, seizures, arrhythmias, AKI (calcium-phosphate precipitation in tubules)
- Hyperuricemia: AKI (uric acid nephropathy), oliguria, flank pain
- AKI: oliguria progressing to anuria, fluid overload
Risk Stratification
| Risk | Features |
|---|---|
| High | Burkitt's lymphoma, ALL with high WBC (>100,000), bulky chemo-sensitive tumors, elevated uric acid/LDH at baseline, renal impairment |
| Intermediate | Most other high-grade lymphomas/leukemias |
| Low | Solid tumors, low-grade hematologic malignancies |
Management (Prevention and Treatment)
| Intervention | Detail |
|---|---|
| Aggressive IV hydration | 0.9% NaCl at 2-3 L/m²/day; maintain urine output >100 mL/h; avoids calcium-phosphate precipitation |
| Allopurinol | Xanthine oxidase inhibitor; prophylaxis for intermediate/high risk; 300 mg/day oral; prevents new uric acid formation but does NOT reduce existing uric acid |
| Rasburicase (recombinant urate oxidase) | Converts uric acid to soluble allantoin; treatment for elevated uric acid or high-risk patients; rapidly lowers uric acid; contraindicated in G6PD deficiency (causes methemoglobinemia and hemolytic anemia) |
| Febuxostat | Xanthine oxidase inhibitor (alternative to allopurinol) |
| Bicarbonate alkalinization | Controversial; increases uric acid solubility but promotes calcium-phosphate precipitation |
| Hyperkalemia management | Calcium gluconate (cardiac protection), insulin/glucose, sodium bicarbonate, kayexalate, dialysis |
| Hypocalcemia | Treat only if symptomatic (avoid giving calcium if high phosphate - worsens precipitation) |
| Renal replacement therapy (dialysis) | For severe refractory AKI, refractory electrolyte disturbances |
| Urine output monitoring | Strict I&O; avoid nephrotoxins |
- Brenner & Rector's The Kidney, p. 1877-1878
6. Febrile Neutropenia
Definition
- ANC <500/μL (or <1000/μL with predicted nadir <500)
- Temperature >38.3°C (single oral reading) or >38.0°C sustained over 1 hour
Clinical Context
- Most common oncological emergency
- Mortality if untreated: ~50%; with prompt appropriate antibiotics: <5%
- Risk of occult bacteremia is high; may have few localizing signs due to inability to mount inflammatory response
Workup
- Blood cultures ×2 (including from each lumen of central venous catheter if present)
- Urine cultures
- CXR
- Respiratory viral panel (if respiratory symptoms)
- Stool cultures if diarrhea
- Complete physical examination (including skin, mouth, perineum, catheter sites)
Empiric Antibiotic Treatment (Start within 60 minutes)
- Piperacillin-tazobactam 4.5 g IV q6-8h
- Cefepime 2 g IV q8h
- Meropenem or imipenem (if β-lactam allergic or resistant organisms suspected)
- Central venous catheter infection suspected
- Mucositis (high-grade)
- Skin/soft tissue infection
- Pneumonia
- Known MRSA colonization
- Hemodynamic instability (septic shock)
- Fever persists after 4-7 days of antibiotics
- Clinical deterioration
- Suspected invasive fungal infection
- High-risk patients (prolonged neutropenia, hematopoietic stem cell transplant)
- Options: micafungin, caspofungin, voriconazole, amphotericin B
MASCC Risk Score (identifies low-risk patients suitable for oral antibiotics/outpatient management)
7. Hyperleukocytosis and Leukostasis
Definition
- Peripheral blast count >100,000/μL in acute leukemia (particularly AML)
- Frequency: 5-13% in AML, 10-30% in ALL; leukostasis rare in lymphoid leukemia
- High risk in: myelomonocytic AML (FAB M4/M5), 11q13/MLL abnormalities, FLT3 mutations
Pathophysiology
Clinical Features
- CNS: stupor, headache, dizziness, tinnitus, visual disturbances, ataxia, confusion, coma, sudden death; papilledema, retinal hemorrhages
- Pulmonary: dyspnea, hypoxemia, respiratory failure; diffuse bilateral infiltrates on CXR
- Other: leg ischemia, renal vein thrombosis, myocardial ischemia, priapism, bowel infarction
Treatment
- Hydroxyurea (1-3 g orally) - rapidly reduces blast count while workup proceeds
- Urgent induction chemotherapy once diagnosis established
- Leukapheresis - for symptomatic leukostasis; temporizing measure
- Avoid RBC transfusions (increases viscosity) and volume depletion
- Monitor for DIC and TLS (common co-complications during induction)
- Avoid platelet transfusions (may worsen leukostasis)
- Harrison's 22E, p. 635
8. Hypercalcemia of Malignancy
Mechanisms
- PTHrP (parathyroid hormone-related protein) secretion - most common (~80%); "humoral hypercalcemia of malignancy"; squamous cell cancers, renal, breast
- Osteolytic metastases - direct bone destruction; breast, myeloma
- Calcitriol (1,25-OH₂ vitamin D) production - lymphoma
- Ectopic PTH secretion (rare)
Clinical Features (Mnemonic: Bones, Stones, Groans, Psychic Moans)
- Bones: bone pain, pathological fractures
- Stones: nephrolithiasis, nephrogenic diabetes insipidus, polyuria, polydipsia
- Groans: nausea, vomiting, constipation, anorexia, pancreatitis
- Psychic moans: confusion, lethargy, depression, psychosis, coma
- Cardiac: short QT interval, arrhythmias, heart block
Treatment
| Severity | Calcium | Treatment |
|---|---|---|
| Mild (asymptomatic) | <12 mg/dL | Hydration, ambulation |
| Moderate/Severe | ≥12 mg/dL or symptomatic | IV hydration + bisphosphonate |
- Aggressive IV saline hydration (3-6 L/day): first-line; promotes renal calcium excretion
- Bisphosphonates (zoledronic acid 4 mg IV over 15 min or pamidronate 60-90 mg IV): onset 24-48h, duration 2-4 weeks; check creatinine first
- Denosumab (RANK-L inhibitor, 120 mg SC): preferred if CrCl <30 mL/min or refractory to bisphosphonates
- Calcitonin (4-8 IU/kg SC/IM q6-12h): rapid onset (hours), mild effect, tachyphylaxis within 48h; useful bridge while bisphosphonates take effect
- Glucocorticoids: for lymphoma/myeloma (calcitriol-mediated)
- Furosemide: only after adequate hydration, use cautiously
- Dialysis: refractory severe hypercalcemia with renal failure
- Treat underlying malignancy
9. SIADH (Syndrome of Inappropriate ADH Secretion)
Causes in Cancer
- SCLC (most common cancer cause - ectopic ADH production)
- CNS tumors, meningeal metastases, brain irradiation
- Drugs: cyclophosphamide, vincristine, cisplatin (vomiting-associated)
- Pulmonary infections, hypothyroidism (non-malignant concurrent causes)
Clinical Features
Treatment
- Mild: fluid restriction (800-1000 mL/day)
- Moderate/Severe: 3% NaCl (hypertonic saline) - correct at max 0.5-1 mEq/L/hour (do not exceed 8-12 mEq/L in 24h - risk of central pontine myelinolysis)
- Tolvaptan (vasopressin V2 receptor antagonist): for refractory cases; monitor Na closely
- Demeclocycline: alternative; causes nephrogenic DI
- Treat underlying malignancy
10. Neoplastic Meningitis (Leptomeningeal Disease)
Causes
- Melanoma, breast (lobular/triple-negative), lung (NSCLC with EGFR/ALK), lymphoma, acute leukemia
- Incidence 3-8% of cancer patients
Clinical Features
- Multifocal neurological signs/symptoms: headache, gait abnormality, mental changes, nausea, vomiting, back/radicular pain, seizures
- Cranial nerve palsies (CN VI, VII most common), limb weakness, paresthesias, decreased DTRs
Investigations
- LP (CSF): malignant cells on cytology (40% false-negative on first tap - repeat at least once); elevated protein; low glucose; elevated CSF pressure
- Gadolinium-enhanced MRI brain + spine (full neuroaxis): leptomeningeal/subependymal/dural/cranial nerve enhancement; intradural nodules; communicating hydrocephalus; 20% have cord abnormalities
- CSF flow studies: abnormal in 70%; important for intrathecal chemotherapy planning
Treatment
- Intrathecal chemotherapy: methotrexate, cytarabine, or thiotepa via lumbar puncture or Ommaya reservoir
- Systemic chemotherapy: CNS-penetrant agents (e.g., high-dose methotrexate, temozolomide, targeted therapy for EGFR/ALK mutations in NSCLC)
- Focal RT: for bulky disease, symptomatic or obstructive lesions, cranial nerve involvement
- VP shunt: for hydrocephalus
- Prognosis: poor (median survival 10-12 weeks); leukemia/lymphoma may be cured if systemic disease controlled
- Harrison's 22E, p. 634
11. Airway Obstruction
Causes
Management
- Humidified oxygen, glucocorticoids
- Heliox (helium-oxygen mixture): reduces turbulent flow, buys time
- Proximal obstruction (larynx/trachea): emergency tracheostomy
- Intrinsic distal obstruction: bronchoscopy + laser (Nd:YAG), argon plasma coagulation, electrocautery, mechanical debulking, intraluminal stenting, photodynamic therapy
- Extrinsic compression: endobronchial stenting, radiation therapy
- Resectable primary airway tumors: surgery
- Harrison's 22E, p. 635
12. Disseminated Intravascular Coagulation (DIC) in Cancer
Common Triggers
- Acute promyelocytic leukemia (APL/AML-M3) - most classic
- Mucin-secreting adenocarcinomas (pancreatic, gastric, lung)
- Massive tumor lysis, sepsis
Treatment
- APL: all-trans retinoic acid (ATRA) is definitive; ATRA itself can be a trigger - treat underlying leukemia promptly
- FFP (clotting factor replacement), cryoprecipitate (fibrinogen/Factor VIII), platelet transfusions
- Heparin: controversial; may be used in chronic DIC from solid tumors
Quick Reference: Key Points per Emergency
| Emergency | Key Diagnostic Clue | Don't Miss |
|---|---|---|
| SVCS | Facial/neck swelling + collateral veins + supine worsening | Airway emergency; get histology before treating |
| Spinal cord compression | Back pain worsening supine + motor/sensory deficits | Start dexamethasone immediately; urgent MRI |
| Cardiac tamponade | Beck's triad + pulsus paradoxus + echo | Immediate pericardiocentesis if hemodynamic instability |
| TLS | Hyperkalemia + hyperphosphatemia + hypocalcemia + hyperuricemia | G6PD deficiency before rasburicase; start prophylaxis before chemo |
| Febrile neutropenia | ANC <500 + T >38.3°C | Antibiotics within 60 minutes; anti-pseudomonal coverage first |
| Hypercalcemia | Polyuria + confusion + short QT | IV saline first; then bisphosphonate |
| Hyperleukocytosis | Blasts >100,000 + respiratory/CNS symptoms | Use pulse oximetry (ABG spurious); avoid transfusion; hydroxyurea + leukapheresis |
| SIADH | Hyponatremia + euvolemia + low serum osmolality | Correct Na slowly (risk central pontine myelinolysis) |
| Neoplastic meningitis | Multifocal CNS symptoms in cancer patient | Repeat CSF if first cytology negative |
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 80 - Oncologic Emergencies (primary comprehensive source)
- The Washington Manual of Medical Therapeutics, Chapter 22
- Brenner & Rector's The Kidney, Chapter 42 (Tumor Lysis Syndrome)
- Tintinalli's Emergency Medicine, Chapter - Oncologic Emergencies
- Wright K et al. Malignant Superior Vena Cava Syndrome: A Scoping Review. J Thorac Oncol 2023. [PMID 37146753]
- Maciel CB & Busl KM. Neuro-oncologic Emergencies. Continuum 2024. [PMID 38830073]
Thyroiditis clinical features diagnosis and management
Thyroiditis - Clinical Features, Diagnosis, and Management
Classification Overview
| Type | Onset | Pain | Thyroid Function | Key Etiology |
|---|---|---|---|---|
| Acute (Suppurative) | Acute | Severe | Usually normal | Bacterial infection |
| Subacute (de Quervain's) | Subacute | Severe, exquisite | Triphasic: hyper → hypo → normal | Viral/post-viral |
| Silent / Postpartum | Subacute | Absent | Triphasic (milder) | Autoimmune |
| Drug-induced | Variable | Usually absent | Variable | Drugs (ICI, IFN-α, amiodarone) |
| Hashimoto's (Chronic lymphocytic) | Insidious/Chronic | Absent | Hypo (or transient hyper) | Autoimmune |
| Riedel's (Fibrous) | Insidious/Chronic | Absent | Usually normal → hypo | Fibro-inflammatory/IgG4 |
1. Acute (Suppurative) Thyroiditis
Epidemiology and Etiology
- Rare (<1% of all thyroid disease); most common in children and young adults
- Most common predisposing condition: pyriform sinus fistula (4th branchial pouch remnant, predominantly left-sided)
- In the elderly: long-standing goiter or degeneration in thyroid malignancy
- Immunocompromised patients (HIV): atypical organisms - fungi, mycobacteria, Pneumocystis
- Common organisms: Staphylococcus aureus and Streptococcus pyogenes (~40% of adults); α- and β-hemolytic streptococcus and anaerobes (children)
Clinical Features
- Thyroid pain - often referred to the throat or ears
- Small, tender, asymmetric goiter
- Fever, dysphagia, erythema over thyroid
- Systemic illness: malaise, cervical lymphadenopathy
- Rapid onset rarely causes diagnostic confusion
Investigations
- Elevated ESR and leukocytosis (left shift)
- Thyroid function usually normal
- FNA biopsy: PMN infiltration; culture identifies organism
- Ultrasound/CT: identifies abscess formation; localizes anatomy
- Thyroid function tests rarely diagnostic
Management
- Antibiotics guided by Gram stain, then cultures from FNA
- Abscess drainage: percutaneous (image-guided) or surgical
- Surgery for abscess or if malignancy cannot be excluded
- Treat predisposing cause (e.g., piriform sinus fistula excision)
- Complications (uncommon with prompt treatment): tracheal obstruction, septicemia, retropharyngeal abscess, mediastinitis, jugular venous thrombosis
- Does not typically result in long-term hypothyroidism
2. Subacute (Granulomatous / de Quervain's) Thyroiditis
Epidemiology
- Most common cause of a painful thyroid gland
- Accounts for up to 5% of clinical thyroid disorders
- Peak incidence: 4th-5th decade; female:male = 3-5:1
- Occurs seasonally (peaks with enterovirus season)
Etiology and Pathophysiology
- Viral or post-viral inflammatory disorder (mumps, coxsackie, influenza, adenovirus, echovirus); also reported after SARS-CoV-2 infection and COVID-19 vaccination
- Pathology: Patchy infiltrate with follicular disruption → multinucleated giant cells → granulomas → fibrosis → recovery
- Triphasic thyroid function due to follicular destruction:
- Thyrotoxic phase: stored hormone leaks from damaged follicles → ↑T4/T3, ↓TSH, LOW radioiodine uptake (<5%)
- Hypothyroid phase: stores depleted; gland not yet recovered → ↓T4, ↑TSH
- Recovery phase: TSH-driven regeneration → normalization
Clinical Features
- Presentation typically 2-3 weeks after upper respiratory illness
- Neck pain, exquisitely tender thyroid - often radiates to jaw, ear, or anterior chest
- Pain may start unilateral and migrate to opposite lobe
- Fever, malaise, myalgias, fatigue
- ~50% of patients have symptoms of thyrotoxicosis during initial phase
- Complete resolution is the usual outcome
- Permanent hypothyroidism in 15% (especially those with coincidental thyroid autoimmunity)
- Prolonged course with relapses in a small percentage
Investigations
| Test | Finding |
|---|---|
| ESR | Markedly elevated - hallmark of subacute thyroiditis during painful phase |
| WBC | Normal or mildly elevated |
| TSH | ↓ (thyrotoxic phase) → ↑ (hypothyroid phase) → normal |
| Free T4/T3 | ↑ (thyrotoxic phase); T4:T3 ratio lower than in Graves' (reflects stored hormone ratio) |
| Radioiodine uptake (RAIU) | Very low or undetectable (<5%) during thyrotoxic phase |
| Thyroid antibodies (TPO, TgAb) | Negative (distinguishes from autoimmune thyroiditis) |
| Thyroid ultrasound | Hypoechoic regions (not routinely required) |
| FNA | Giant cells, granulomas; useful if unilateral to exclude hemorrhage or neoplasm |
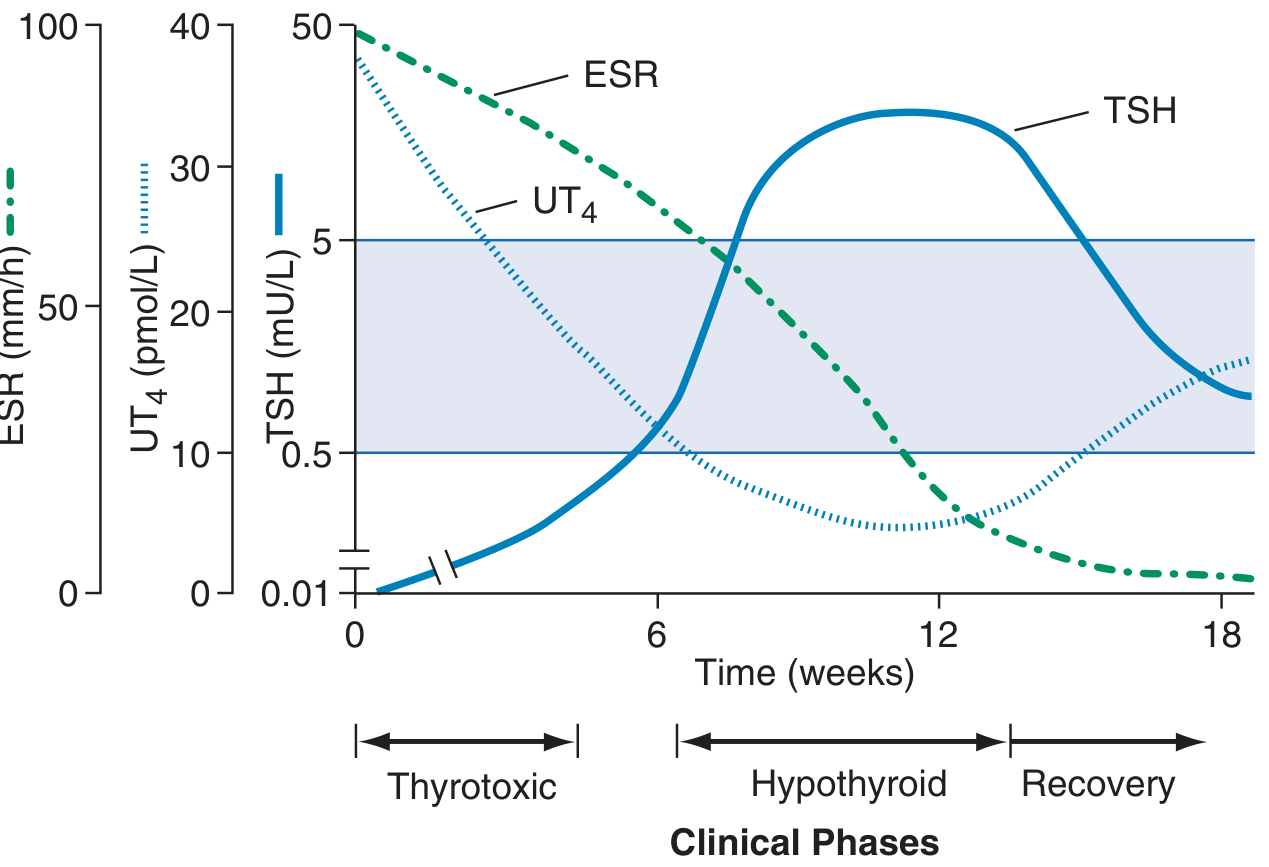
Management
- Mild-moderate pain: NSAIDs (e.g., aspirin 600 mg q4-6h) + PPI gastroprotection
- Severe symptoms or inadequate NSAID response: Oral prednisone 15-40 mg/day (taper over 6-8 weeks guided by symptoms and ESR); if relapse during taper, increase dose and taper more slowly
- Thyrotoxic symptoms: β-blockers (propranolol 20-40 mg tid-qid or atenolol 25-50 mg daily) - symptomatic only; antithyroid drugs (PTU/methimazole) are NOT indicated (synthesis is not increased - hormone is released from damaged follicles)
- Hypothyroid phase: Levothyroxine replacement (50-100 μg daily) if prolonged or symptomatic; use low enough dose to allow TSH-mediated recovery
- Monitoring: TSH and free T4 every 2-4 weeks
3. Silent (Painless) Thyroiditis and Postpartum Thyroiditis
Definition and Epidemiology
- Painless thyroiditis = silent thyroiditis; postpartum thyroiditis = occurs within 12 months after delivery (essentially the same disorder)
- Postpartum thyroiditis: up to 10% of pregnancies; recurs after ~70% of subsequent pregnancies
- 3× more common in women with type 1 diabetes mellitus
- More frequent with personal/family history of autoimmunity
- In the postpartum setting, 20× more common than Graves' hyperthyroidism
Pathogenesis
- Autoimmune destruction-induced thyroiditis
- Associated with positive TPO antibodies (present antepartum)
- Unlike subacute thyroiditis, the immune response CAN be self-perpetuating → risk of permanent hypothyroidism
Clinical Features (Classic Triphasic Sequence)
- Thyrotoxic phase (1-3 months): tachycardia, palpitations, heat intolerance, nontender goiter; symptoms milder than Graves' disease; substantial weight loss is rare
- Brief euthyroid period (1-2 months) (may be absent)
- Hypothyroid phase (up to 12 months): fatigue, impaired concentration, constipation, cold intolerance
- ~25% have only the thyrotoxic phase; ~50% have only the hypothyroid phase
- Some patients never develop clinically apparent thyrotoxicosis
Investigations
| Test | Finding |
|---|---|
| TSH | ↓ (thyrotoxic) → ↑ (hypothyroid) |
| Free T4/T3 | Elevated in thyrotoxic phase; T3:T4 ratio <20 (vs. Graves') |
| ESR | Normal (key differentiator from subacute thyroiditis) |
| TPO antibodies | Positive in most patients |
| TSH receptor antibodies (TRAb) | Negative (distinguishes from Graves') |
| RAIU | Low/undetectable during thyrotoxic phase |
| Thyroid ultrasound | Nontender goiter; generalized hypoechogenicity |
| Clinical | No exophthalmos, no thyroid bruit (absent stigmata of Graves') |
Management
- Thyrotoxic phase: β-blockers (atenolol 25-50 mg daily) until hormone levels normalize; antithyroid drugs NOT useful (synthesis not increased)
- Hypothyroid phase: Levothyroxine if symptomatic or if pregnant/attempting conception; taper after 6-12 months to assess recovery
- Long-term: Annual thyroid function follow-up; up to 50% develop permanent hypothyroidism (higher risk with: high-titer TPO antibodies, missed thyrotoxic phase, severe hypothyroidism)
- Glucocorticoids NOT indicated in silent thyroiditis
4. Drug-Induced Thyroiditis
Causes
- Immune checkpoint inhibitors (ICIs): pembrolizumab, nivolumab - thyroiditis in 5-20% of treated cancer patients; painless thyroiditis pattern
- Interferon-α (IFN-α): used in hepatitis B/C, hematologic malignancies; thyroid dysfunction in up to 5%; commonest in women with pre-existing TPO antibodies; can cause painless thyroiditis, hypothyroidism, or Graves' disease
- Tyrosine kinase inhibitors (TKIs): sorafenib and others
- Amiodarone: can cause thyroiditis or thyrotoxicosis (two distinct types: iodine-excess thyrotoxicosis = type 1; destructive thyroiditis = type 2); may be acute, subacute, or chronic
Management
- Same approach as silent thyroiditis: β-blockers for thyrotoxicosis, levothyroxine for hypothyroidism
- ASCO recommends routine TFT monitoring during ICI therapy
- For amiodarone-induced destructive thyroiditis (type 2): glucocorticoids (prednisone 40 mg/day); consider discontinuation of amiodarone if clinically feasible
5. Hashimoto's Thyroiditis (Chronic Lymphocytic Thyroiditis)
Epidemiology
- Most common cause of hypothyroidism in iodine-sufficient regions of the world
- Most prevalent 45-65 years; female:male = 10-20:1
- Strong genetic component: CTLA4 polymorphisms; 40% concordance in monozygotic twins; 50% of asymptomatic siblings have antithyroid antibodies
Pathogenesis (Autoimmune Destruction)
- Multiple immunological mechanisms:
- CD8+ cytotoxic T cells kill thyroid epithelial cells
- CD4+ Th1 cells release IFN-γ → macrophage activation → follicular destruction
- Anti-TPO and anti-thyroglobulin antibodies → antibody-dependent cytotoxicity and complement activation
Pathology (Robbins)
- Diffuse, symmetric thyroid enlargement
- Dense lymphocytic infiltrate with prominent germinal centers
- Hürthle (oxyphil) cells - follicular cells with abundant eosinophilic granular cytoplasm (mitochondria-rich metaplasia in response to injury)
- Atrophic follicles; increased fibrous stroma
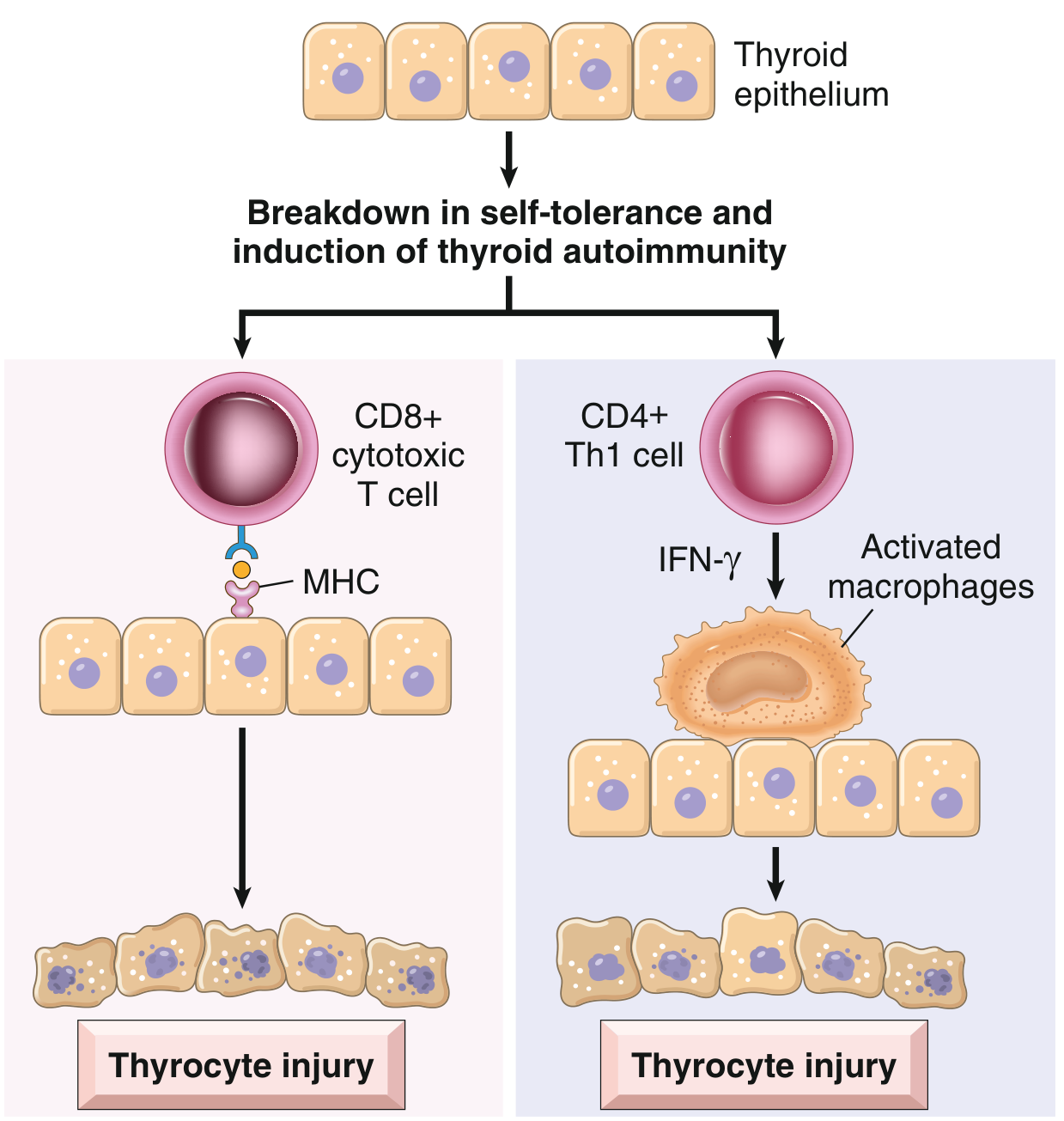
Clinical Features
- Painless, diffuse, firm/rubbery goiter of variable size - most common presentation
- Slowly progressive hypothyroidism (most common clinical outcome): fatigue, cold intolerance, weight gain, constipation, dry skin, hair loss, bradycardia, periorbital puffiness
- Hashitoxicosis: transient thyrotoxicosis due to follicular disruption releasing stored hormone; precedes hypothyroidism in some patients; elevated T4/T3, suppressed TSH, decreased RAIU
- Associated with other autoimmune conditions (Type 1 DM, vitiligo, pernicious anemia, Addison's disease, Sjögren's, lupus)
- Increased risk of B-cell non-Hodgkin lymphoma (primary thyroid lymphoma)
- Possible increased risk of papillary thyroid carcinoma (controversial)
- Focal thyroiditis: present in 20-40% of euthyroid patients at autopsy (subclinical autoimmunity)
Investigations
| Test | Finding |
|---|---|
| TSH | Elevated (hypothyroidism) |
| Free T4 | Low or low-normal |
| Anti-TPO antibodies | Positive in >90% - most sensitive marker |
| Anti-thyroglobulin (TgAb) | Positive in ~60% |
| TSH receptor antibodies | Negative |
| Thyroid ultrasound | Diffuse hypoechoic, heterogeneous parenchyma; often hypervascular; may show pseudo-nodules |
| FNA biopsy | Lymphocytic infiltration, Hürthle cell change, germinal centers (rarely needed unless nodule suspicious) |
Management
- Confirmed hypothyroidism: Levothyroxine (LT4) replacement - starting dose 1.6 μg/kg/day; titrate to normalize TSH; check TSH 6-8 weeks after dose change
- Subclinical hypothyroidism (elevated TSH, normal T4): treat if TSH >10 mIU/L, or if symptomatic, or if pregnant (or attempting)
- Euthyroid Hashimoto's: no treatment needed; monitor TSH annually
- Goiter causing compressive symptoms: LT4 suppression rarely effective; surgical resection if significant compression
- Selenium supplementation: may reduce TPO antibody titers (evidence limited)
- Annual monitoring: TFTs; screen for associated autoimmune conditions
- Thyroid lymphoma suspected: urgent CT + FNA/core biopsy
6. Riedel's Thyroiditis (Invasive Fibrous Thyroiditis)
Overview
- Rare, poorly understood fibroinflammatory disorder causing progressive fibrosis of the thyroid with extension into surrounding structures
- Predominantly affects middle-aged women
- Strong association with IgG4-related disease (idiopathic fibrosis at multiple sites: retroperitoneum, mediastinum, biliary tree, lung, orbit)
Clinical Features
- Insidious, painless goiter
- Rock-hard, fixed, non-tender thyroid - clinically mimics malignancy (anaplastic carcinoma, lymphoma)
- Compressive symptoms due to fibrosis extending beyond thyroid capsule:
- Dysphagia (esophageal compression)
- Dyspnea/stridor (tracheal compression)
- Dysphonia/hoarseness (recurrent laryngeal nerve involvement)
- Venous congestion (neck vein compression)
Investigations
- TSH/T4: hypothyroidism when significant gland destruction; may be normal early
- Ultrasound: diffusely hypoechoic with ill-defined borders
- CT/MRI: defines extent; evaluates tracheal narrowing
- FNA: dense fibrotic changes - cannot reliably exclude anaplastic thyroid carcinoma
- Open (surgical) biopsy required for definitive diagnosis (FNA inadequate)
- Serum IgG4 levels may be elevated
Management
- Medical (inflammatory component):
- Glucocorticoids (prednisone): first-line anti-inflammatory
- Tamoxifen (antifibrotic): widely used; may have independent benefit via TGF-β suppression
- Other agents: mycophenolate mofetil, rituximab
- Hypothyroidism: levothyroxine replacement
- Surgery: indicated for:
- Exclude malignancy (ATC or thyroid lymphoma cannot be excluded by FNA)
- Relieve aerodigestive tract compression (isthmus wedge resection most common)
- Surgery is technically challenging due to obliterated tissue planes; only partial resection recommended
- Not associated with increased risk of thyroid malignancy
Differential Diagnosis: Painful vs. Painless Thyroiditis
| Feature | Subacute (de Quervain's) | Silent/Postpartum | Hashimoto's | Acute Suppurative |
|---|---|---|---|---|
| Pain | Severe, exquisite | Absent | Absent | Severe |
| Fever | Common | Absent | Absent | High fever |
| ESR | Very high | Normal | Normal | Elevated |
| WBC | Normal/mildly elevated | Normal | Normal | Leukocytosis |
| TPO antibodies | Usually negative | Positive | Positive (>90%) | Negative |
| RAIU (thyrotoxic phase) | Low (<5%) | Low | Low (Hashitoxicosis) | Normal |
| T3:T4 ratio | <20 (stored hormone) | <20 | Variable | Normal |
| TRAb | Negative | Negative | Negative | Negative |
| Response to steroids | Yes | No | No | No |
Key Distinguishing Point: Painless Thyrotoxicosis
- Silent/postpartum thyroiditis
- Subacute thyroiditis (if painful)
- Drug-induced thyroiditis (ICI, IFN-α)
- Factitious thyrotoxicosis (exogenous T4/T3 ingestion - also low RAIU, low Tg)
- Struma ovarii (ectopic thyroid tissue)
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 396 - Thyroiditis (primary source, all types)
- Goldman-Cecil Medicine, Chapter 207 - Postpartum and Sporadic Painless Thyroiditis; Subacute Thyroiditis
- Robbins & Kumar Basic Pathology (Robbins Pathology) - Hashimoto's and de Quervain's pathology and morphology
- Sabiston Textbook of Surgery - Riedel's thyroiditis and acute suppurative thyroiditis