Encephalopathy
Reading File
Checking Date & Time
Asking for Preferences
Finding Sources
Searching PubMed
encephalopathy classification pathophysiology management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Here is a structured overview covering all major types of encephalopathy at a detailed clinical level.
Encephalopathy: Comprehensive Clinical Review
Definition
Encephalopathy is diffuse brain dysfunction caused by a systemic, metabolic, toxic, or inflammatory insult rather than primary intrinsic brain disease. It manifests as a spectrum from mild inattention → confusion → stupor → coma. Phylogenetically newer brain structures (cortex, complex polysynaptic pathways) are affected first; brainstem reflexes are preserved until near-terminal stages. — Localization in Clinical Neurology, 8e
General Clinical Features of Metabolic Encephalopathy
| Sign | Details |
|---|---|
| Asterixis | Sudden, brief loss of postural tone ("flapping tremor"); elicited by dorsiflexing wrist with fingers extended; present in mild–moderate encephalopathy, disappears in deep coma |
| Tremor | Coarse, irregular, 8–10 Hz; maximal with outstretched hands |
| Multifocal myoclonus | Non-rhythmic muscle twitching; seen in uremic, hyperosmolar-hyperglycaemic, and CO₂ narcosis |
| Hyperventilation / respiratory alkalosis | Prominent in hepatic and septic encephalopathy |
| EEG slowing | Correlates with clinical severity |
Asymmetric motor signs argue against a purely metabolic cause — consider concurrent structural lesion.
I. Hepatic Encephalopathy (HE)
Epidemiology
Develops in 50–70% of cirrhotic patients; onset signals poor prognosis (1-year survival 42%, 3-year survival 23% without liver transplantation).
Classification
By underlying disease:
- Type A — acute liver failure (ALF)
- Type B — portosystemic bypass without liver disease
- Type C — cirrhosis + portal hypertension (most common)
By severity — West Haven Criteria & SONIC Classification:
| Grade | Cognition | Neuromuscular | SONIC Category |
|---|---|---|---|
| 0 | Normal | Normal | Unimpaired |
| Minimal | Subtle work/driving changes | Minor visuospatial abnormalities | Covert HE |
| 1 | Shortened attention, mild confusion | Tremor, incoordination | Covert HE |
| 2 | Lethargy, disorientation to time | Asterixis, ataxia | Overt HE |
| 3 | Gross disorientation, somnolence | Asterixis, rigidity | Overt HE |
| 4 | Coma | Decerebrate posturing | Overt HE |
Pathophysiology
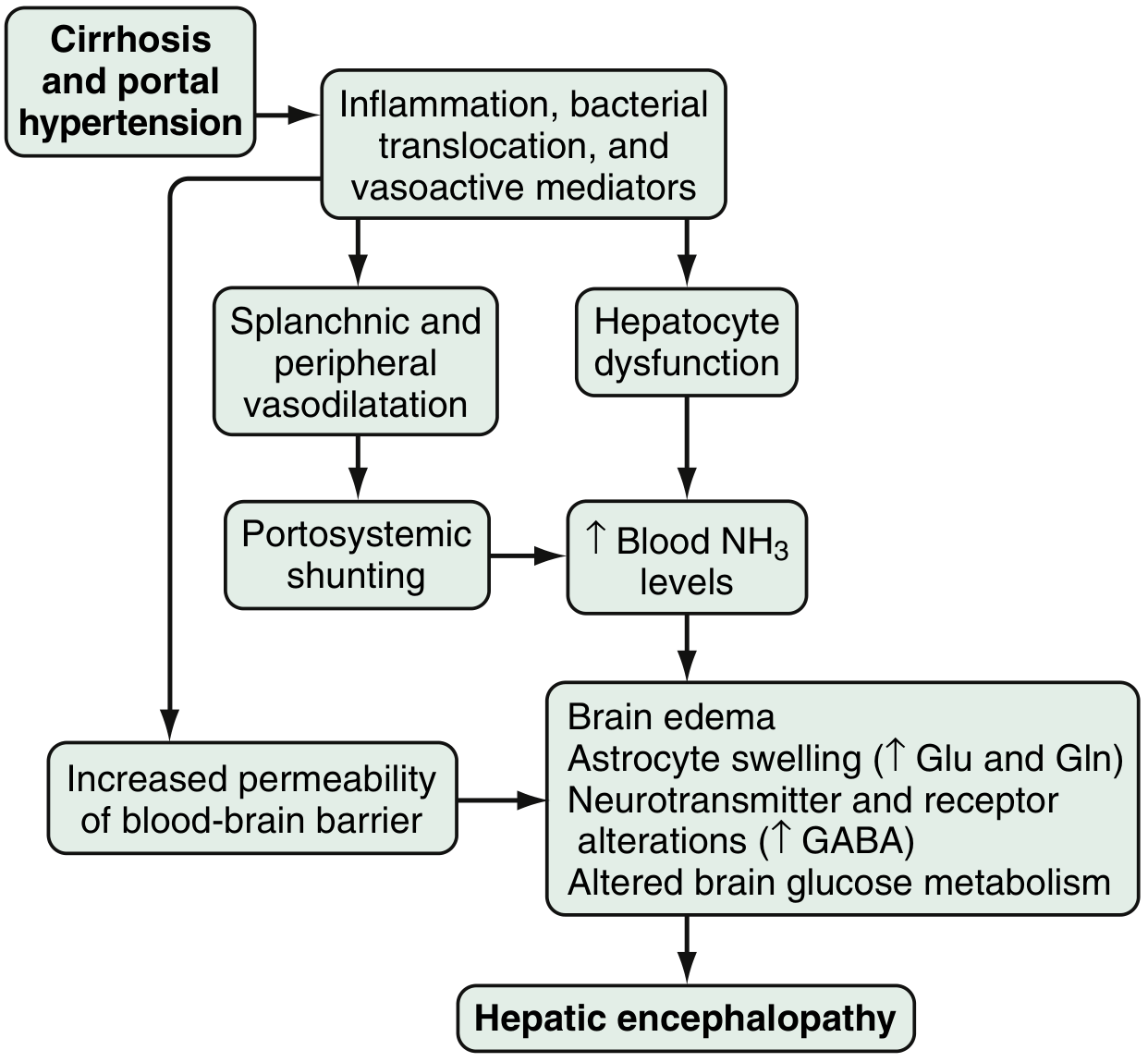
- Hyperammonemia — ammonia from colonic bacteria/enterocyte glutamine metabolism bypasses the liver via portosystemic shunting → crosses BBB as NH₃ gas → astrocytes convert it to glutamine → astrocyte swelling, cytotoxic edema. Arterial hyperammonemia present in ~90% of HE cases, but serum levels are neither sensitive nor specific.
- Astrocyte swelling — the key pathological event; amplified by inflammatory cytokines, hyponatraemia, and benzodiazepines (explaining why these precipitate HE). Depletion of intracellular myoinositol (which normally counteracts swelling) increases vulnerability.
- GABA-benzodiazepine system — enhanced astrocyte peripheral benzodiazepine receptor sensitivity → ↑ neurosteroid production (allopregnanolone, THDOC) → amplified GABAergic inhibition.
- Neuroinflammation — intercurrent infection drastically worsens HE by direct cytokine-mediated brain dysfunction.
- Other: manganese (dopaminergic toxicity), nitric oxide, reactive oxygen species, gut microbiome dysbiosis.
Common Precipitants
GI bleeding · hypokalemia · infection (SBP, UTI, pneumonia) · dehydration · constipation · excess dietary protein · sedatives/opioids · TIPS placement
Clinical Features
- Early: forgetfulness, sleep-wake reversal, changes in handwriting, difficulty driving
- Progression: asterixis → agitation → disinhibited behaviour → seizures → coma
- Typical onset: quiet, apathetic delirium (10–20% present with agitated/manic delirium)
- Respiratory alkalosis with hyperventilation is nearly invariable — its absence argues against hepatic coma
- Pupillary and caloric responses remain normal until preterminal stages (distinguishes from structural brainstem lesions) — Plum & Posner
Management
| Intervention | Details |
|---|---|
| Identify and treat precipitants | First and most critical step |
| Lactulose | 15–45 mL PO/NG BID–QID; titrate to 2–3 soft stools/day; acidifies colon, traps NH₃ as NH₄⁺ |
| Rifaximin | 400 mg q8h or 550 mg BID; non-absorbable antibiotic; superior to lactulose alone in several RCTs; used for secondary prophylaxis |
| Branched-chain amino acids | IV infusion; proven benefit without increased mortality |
| Protein management | Moderate restriction; avoid excess — muscle is the main extra-hepatic ammonia detoxifier |
| Liver transplantation | Definitive; generally reverses HE |
In ALF (Type A), intracranial hypertension develops in up to 80% of grades 3–4 encephalopathy. ICP monitoring, mannitol, and hypertonic saline may be needed.
II. Uremic Encephalopathy
Clinical Features
- Early: lethargy, impaired attention/memory (~30% of dialysis patients have neuropsychiatric symptoms)
- Intermediate: confusion, hallucinations, psychosis, tremor, myoclonus, asterixis
- Severe: tonic-clonic or myoclonic seizures, stupor, coma
- Degree of azotaemia correlates poorly with severity — urea itself is not the neurotoxin
Pathophysiology
- Guanidine compounds (guanidinusuccinic acid, methylguanidine) — elevated 100-fold in uremic brain/CSF; activate NMDA receptors → seizures
- Cytokine-mediated neuroinflammation + ↑ BBB permeability
- Secondary hyperparathyroidism → elevated brain calcium → disrupted neurotransmitter release
- Gut-microbial metabolites (phenylalanine, benzoate, glutamate pathways) linked to cognitive impairment
- Anaemia — EPO therapy improves cognitive performance
Related Dialysis Syndromes
| Syndrome | Mechanism | Features |
|---|---|---|
| Dialysis disequilibrium | Rapid solute clearance → osmotic gradient → cerebral edema | Headache, confusion, seizures during/after HD; treat by reducing dialysis duration/frequency |
| Dialysis encephalopathy | Aluminium neurotoxicity (historical) | Dysarthria, aphasia, myoclonus, cognitive decline |
| PRES | Failed autoregulation + endothelial dysfunction + fluid overload | Headache, visual changes, seizures; posterior white-matter edema on MRI |
| Wernicke's (dialysis) | Thiamine loss via dialysis; 33% prevalence in symptomatic dialysis encephalopathy | Classic triad (see below) |
Management
- Renal replacement therapy (dialysis/transplantation) — definitive
- Correct anaemia and hyperparathyroidism
- PRES: volume control and BP management
- Caution with AEDs: uraemia + hypoalbuminaemia alter phenytoin protein binding; free levels should be measured directly
III. Hypertensive Encephalopathy & PRES
Hypertensive Encephalopathy
Mechanism: autoregulatory failure → breakthrough hyperperfusion → vasospasm → ischemia → ↑ vascular permeability → vasogenic oedema + punctate haemorrhages
Clinical: severe headache, vomiting, altered mental status, seizures/coma; visual disturbance → blindness; papilloedema; hypertensive retinopathy. Focal deficits do not follow a single anatomic distribution (diffuse dysfunction, not stroke).
Diagnosis: clinical (diffuse neurological dysfunction + markedly elevated BP ± papilloedema) + CT showing non-specific or absent changes. Do not delay treatment waiting for imaging.
Management: controlled BP reduction by 30–40% using IV agents (labetalol, nicardipine, nitroprusside). Fully reversible with early treatment; in-hospital mortality <1%. — Rosen's Emergency Medicine
PRES
- Similar pathophysiology, but more posterior and region-specific
- MRI hallmark: vasogenic oedema in posterior parietal-temporal-occipital white matter
- Seizures in up to 90% of PRES cases
- Causes beyond hypertension: eclampsia, vasculitis, TMA, calcineurin inhibitors (ciclosporin, tacrolimus), rituximab, ESKD, erythropoietin therapy
- Reversible with treatment of underlying cause
IV. Wernicke Encephalopathy
Pathophysiology
Thiamine (B₁) deficiency impairs oxidative metabolism in high-demand regions: mammillary bodies, anterior/centromedian thalamus, periaqueductal grey, floor of the fourth ventricle. Deficiency of α-ketoglutarate dehydrogenase in astrocytes → microglial activation → glutamatergic toxicity → neuronal swelling, microscopic haemorrhages, gliosis.
At-Risk Groups
Chronic alcohol abuse · prolonged vomiting (any cause) · bariatric surgery · AIDS/cancer cachexia · dialysis patients · malnutrition
Classic Triad (present in only ~1/3 of cases)
- Mental status change — inattention → delirium → coma; often apathy/abulia
- Oculomotor dysfunction — nystagmus, dysconjugate gaze, gaze palsies
- Ataxia — gait and lower limb predominance; non-alcoholic patients have more ocular involvement
⚠️ Only ~25% of cases are recognised before death. Clinically missed in most patients.
Investigations
- MRI T2/FLAIR: symmetrical hyperintensity at periventricular thalamus, periaqueductal grey, floor of fourth ventricle, mammillary bodies
- Thiamine level <50 μg/mL (normal in ~10%)
- Elevated lactate and pyruvate
⚠️ Give IV thiamine BEFORE any glucose — IV glucose alone can precipitate acute Wernicke's
Management
- IV thiamine (200–500 mg TID) immediately, before glucose
- Followed by oral thiamine supplementation
- Untreated → Korsakoff syndrome (irreversible anterograde amnesia + confabulation)
V. Septic/Systemic Inflammatory Encephalopathy
Mechanisms (converging):
- Cytokine/LPS-mediated BBB disruption
- Altered cerebral microcirculation and mitochondrial dysfunction
- Endotoxin-driven oxidative stress and neurotransmitter disturbances
- Sickness syndrome: PGE₂ crosses BBB → hypothalamic activation (fever, somnolence, anorexia, lowered pain threshold)
Key clinical clue: hyperventilation + respiratory alkalosis + impaired consciousness with no other metabolic cause → mount an urgent search for occult infection (UTI, wound infection). Treating the infection source often reverses the encephalopathy. — Plum & Posner
VI. Radiation Encephalopathy
| Type | Timing | Mechanism | Features | Treatment |
|---|---|---|---|---|
| Acute | Days post-RT | BBB breakdown, ↑ ICP | Headache, nausea, altered mental status | High-dose corticosteroids |
| Early delayed | 2 wks–4 months | Demyelination | Headache, drowsiness, previous deficits enhanced | Self-limiting; no specific treatment |
| Late (delayed) | Months–years (esp. >35 Gy total dose) | Vascular changes, necrosis, neuronal loss | Progressive dementia, frontal dysfunction, gait disorder | Bevacizumab; experimental: stem cell/anti-inflammatory |
Bradley & Daroff's Neurology in Clinical Practice
Diagnostic Approach
All encephalopathy patients:
1. Exclude hypoglycaemia → IV thiamine then glucose if malnourished
2. Structural imaging (CT/MRI) if focal signs, trauma, or anticoagulation
3. Basic labs: glucose, Na, K, Ca, Mg, BUN/Cr, LFTs, ammonia, ABG, TFTs
4. Toxicology screen; medication review
5. Sepsis workup (CBC, cultures, CXR, UA)
6. EEG if subclinical seizures suspected
→ Treat the identified cause:
Glucose → hypoglycaemia
Thiamine → Wernicke's
Lactulose ± rifaximin → hepatic
Dialysis → uremic
BP control → hypertensive/PRES
Antibiotics + source control → septic
Remove offending agent → toxic
Sources: Sleisenger & Fordtran's GI and Liver Disease · Plum & Posner's Diagnosis and Treatment of Stupor and Coma · Bradley & Daroff's Neurology in Clinical Practice · Brenner & Rector's The Kidney · Rosen's Emergency Medicine · Goldman-Cecil Medicine · Localization in Clinical Neurology, 8e
Para neoplastic syndrome
Checking Date & Time
Finding Sources
Searching PubMed
paraneoplastic neurological syndrome autoantibodies
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is the comprehensive review:
Paraneoplastic Syndromes: Comprehensive Clinical Review
Definition
Paraneoplastic syndromes (PNS) are remote effects of cancer on the nervous system (or other organs) that are not caused by direct tumour invasion, metastasis, infection, ischemia, metabolic derangement, or treatment. They are autoimmune disorders where the tumour expresses antigens normally confined to neurons — triggering an immune response that cross-attacks the nervous system (onconeural antigens). — Bradley & Daroff's Neurology
Critical clinical point: neurological symptoms often precede tumour detection by 4–12 months and progress rapidly over weeks to months. Recognising the syndrome early is the key to identifying a potentially treatable cancer.
Pathophysiology: The Two-Subtype Paradigm
The most clinically important distinction is based on where the target antigen is located:
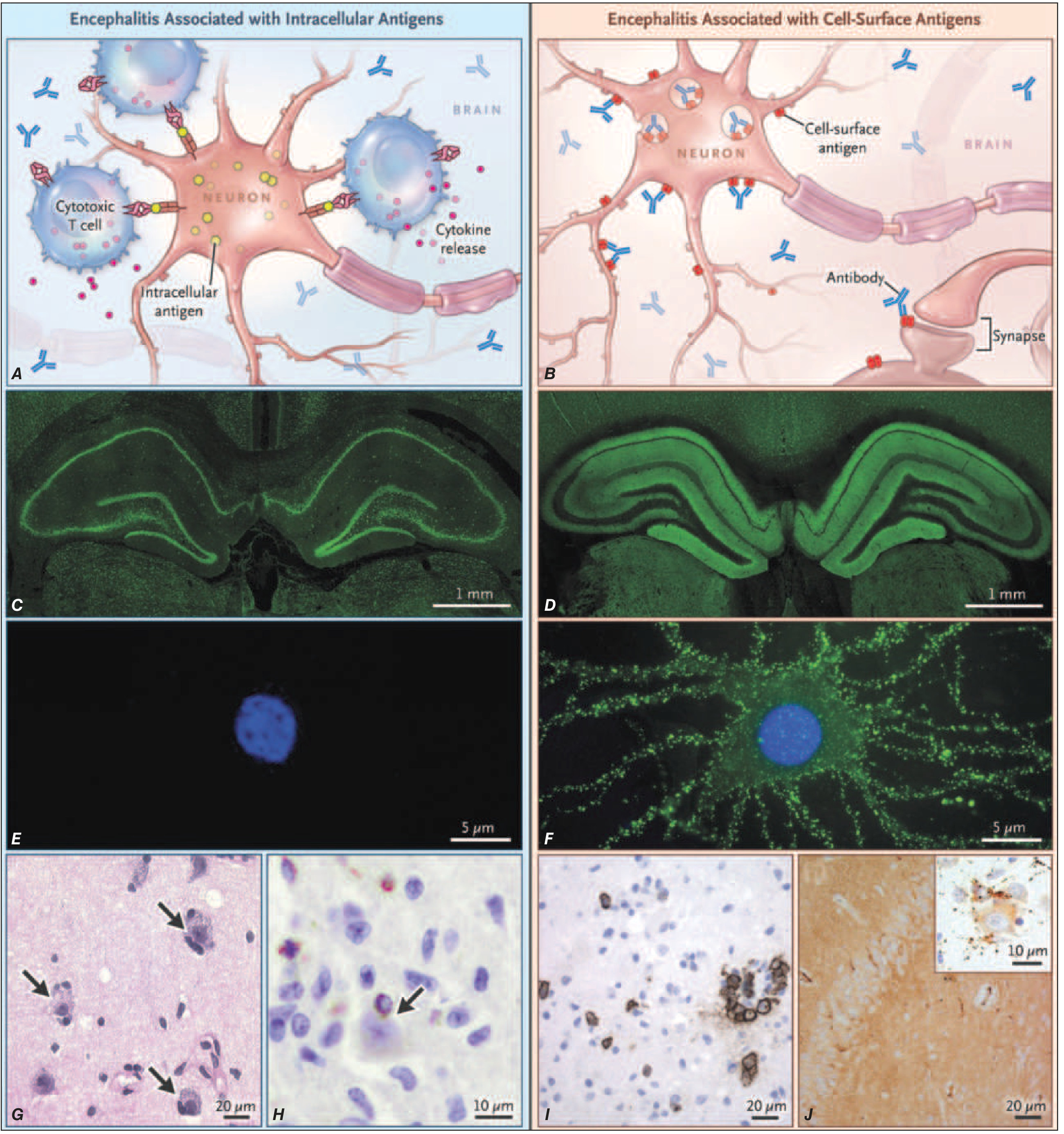
| Feature | Intracellular Antigens | Cell-Surface/Synaptic Antigens |
|---|---|---|
| Examples | Anti-Hu, Anti-Yo, Anti-Ri, Anti-Ma2, Anti-CV2 | Anti-NMDAR, Anti-LGI1, Anti-Caspr2, Anti-AMPAR |
| Effector mechanism | Cytotoxic T cells → irreversible neuronal loss | Antibodies directly alter receptor function → reversible synaptic dysfunction |
| Cancer prediction | Highly predictive (almost always malignancy) | Less predictive; many autoimmune variants without cancer |
| Treatment response | Poor — stabilisation is the goal | Good — often substantial recovery |
Antibody Table and Tumour Associations
Classic Onconeural Antibodies (Intracellular Targets)
| Antibody | Syndrome | Primary Tumour |
|---|---|---|
| Anti-Hu (ANNA-1) | Sensory neuronopathy, PEM, autonomic neuropathy, GI dysmotility | SCLC |
| Anti-Yo (PCA-1) | Subacute cerebellar degeneration | Ovarian, breast |
| Anti-Ri (ANNA-2) | Opsoclonus-myoclonus, jaw dystonia, ataxia | Breast, SCLC |
| Anti-Ma2 (Ta) | Limbic/hypothalamic/brainstem encephalitis | Testicular germ cell (men <45 y); lung |
| Anti-CV2/CRMP5 | Cerebellar ataxia, dementia, chorea, uveitis, sensorimotor neuropathy | SCLC, thymoma |
| Anti-amphiphysin | Stiff-person syndrome, PEM | Breast, SCLC |
Cell-Surface / Synaptic Antibodies
| Antibody | Syndrome | Tumour Association |
|---|---|---|
| Anti-NMDAR | Anti-NMDAR encephalitis (see below) | Ovarian teratoma (~50% in young women) |
| Anti-LGI1 | Limbic encephalitis, faciobrachial dystonic seizures, hyponatraemia | Thymoma <5%; mostly autoimmune |
| Anti-Caspr2 | Morvan syndrome, encephalitis, peripheral nerve hyperexcitability | Thymoma ~20% (Morvan ~50%) |
| Anti-AMPAR | Limbic encephalitis, psychiatric features | Breast, SCLC, thymoma |
| Anti-GABA-B | Limbic encephalitis, refractory seizures | SCLC/neuroendocrine (~50%) |
| Anti-GABA-A | Severe refractory status epilepticus | Thymoma |
| Anti-GlyR | PERM (rigidity + myoclonus) | Thymoma, lymphoma, breast (~20%) |
| Anti-VGCC | LEMS, cerebellar ataxia | SCLC |
Classic Syndromes
1. Paraneoplastic Encephalomyelitis (PEM)
Multifocal neuraxis involvement. Features vary by predominant site:
- Limbic: memory loss, confusion, seizures, psychiatric features
- Brainstem: oscillopsia, diplopia, dysarthria, gaze palsies
- Cerebellar: gait ataxia; Purkinje cell loss with T-cell infiltrates
- Autonomic: postural hypotension, gastroparesis, neurogenic bladder; cardiac arrhythmia/respiratory failure = common causes of death
- Spinal cord (myelitis): lower motor neuron features ~20%
Antibody: Anti-Hu with SCLC (most common). Poorly responsive to treatment.
2. Limbic Encephalitis (LE)
- Clinical: subacute short-term memory loss + complex partial seizures + psychiatric symptoms (confusion, agitation, hallucinations)
- MRI: unilateral or bilateral mesial temporal lobe T2/FLAIR signal increase — one of the few PNS with characteristic imaging
- Antibody clues:
- Anti-LGI1 → faciobrachial dystonic seizures + hyponatraemia + male >50 years → responds well to immunotherapy
- Anti-Ma2 → young male + vertical gaze palsy + testicular tumour → ~1/3 improve with treatment
- Anti-Hu → part of PEM, SCLC → poor prognosis
3. Anti-NMDA Receptor Encephalitis
The most common autoimmune encephalitis. Five-stage progression:
- Prodrome (flu-like)
- Psychiatric phase — hallucinations, delusions, agitation (frequently misdiagnosed as first-episode psychosis)
- Unresponsive/catatonic phase
- Hyperkinetic phase — orofacial dyskinesias, autonomic instability, central hypoventilation
- Recovery (months)
MRI: often normal. Tumour: ovarian teratoma in ~50% of young women; screen with pelvic US/MRI/CT. Children and males often have no tumour. Responds well to immunotherapy + tumour removal.
4. Paraneoplastic Cerebellar Degeneration (PCD)
- Subacute pancerebellar syndrome: truncal + limb ataxia, nystagmus, dysarthria, diplopia
- Accounts for ~12% of paraneoplastic syndromes in lung cancer
- Pathology: massive Purkinje cell loss with CD3 T-cell infiltrates
- Antibodies: Anti-Yo (ovary/breast), Anti-Hu (SCLC), Anti-VGCC (often with LEMS), Anti-Ri
- Prognosis: severe, largely irreversible unless cell-surface antibody involved
5. Paraneoplastic Sensory Neuronopathy (PSN)
- Progressive sensory loss all modalities + painful dysesthesias; initially asymmetric
- Sensory ataxia, pseudoathetotic movements; hearing loss possible
- 2/3 develop symptoms before cancer diagnosis
- NCS: absent/small SNAPs; normal motor studies (dorsal root ganglion pathology)
- Antibody: Anti-Hu (~80%; SCLC)
- Management: tumour control → stabilisation; corticosteroids may partially help
6. Paraneoplastic Opsoclonus-Myoclonus (POM)
- Chaotic, arrhythmic conjugate saccades in all directions + multifocal myoclonus
- Children: neuroblastoma; prominent gait disturbance; treat with ACTH, IVIg
- Adults: breast (anti-Ri), SCLC, testicular; worse course
7. Lambert-Eaton Myasthenic Syndrome (LEMS)
- Mechanism: Anti-VGCC antibodies → impaired presynaptic ACh vesicle release
- Clinical: proximal leg > arm weakness, fatigue, dry mouth; brief exercise temporarily improves strength (Lambert's sign)
- EMG: incremental response on repetitive stimulation (vs. decrement in MG)
- Tumour: SCLC ~60%; if negative, screen 6-monthly for ≥3 years
- Treatment: 3,4-diaminopyridine, pyridostigmine, IVIg, plasma exchange + tumour treatment
8. Stiff-Person Syndrome (Paraneoplastic)
- Progressive axial rigidity + painful spasms on stimulation + lumbar hyperlordosis
- Paraneoplastic: Anti-amphiphysin → breast/SCLC
- Autoimmune variant: Anti-GAD65 (not paraneoplastic)
- Treatment: diazepam, baclofen, IVIg, plasma exchange
9. POEMS Syndrome
Polyneuropathy · Organomegaly · Endocrinopathy · M-protein · Skin changes
Associated with sclerotic myeloma or Castleman disease. Neuropathy mimics CIDP but with more axonal loss; elevated serum VEGF is key. Treatment: high-dose alkylator chemotherapy + autologous stem cell transplantation.
Diagnostic Criteria (Graus et al., 2004) — Any ONE of:
- Classical syndrome + cancer within 5 years (antibody-independent)
- Non-classical syndrome that objectively improves with cancer treatment
- Non-classical syndrome + paraneoplastic antibodies + cancer within 5 years
- Neurological syndrome (any) + well-characterised onconeural antibody
Diagnostic Workup
1. MRI brain ± spine
→ LE: bilateral medial temporal FLAIR ↑
→ PCD: normal early → late cerebellar atrophy
→ Anti-NMDAR: often normal
2. CSF: lymphocytic pleocytosis, ↑protein, oligoclonal bands
→ Rule out CNS infection, leptomeningeal disease
3. Onconeural antibody panel — BOTH serum AND CSF
→ Only 60–70% of CNS PNS are antibody-positive
→ Positive antibody = mandatory tumour search
4. Tumour search
→ CT chest/abdomen/pelvis
→ PET-CT (best sensitivity for occult tumours)
→ Pelvic US/MRI (ovarian teratoma)
→ Testicular US (anti-Ma2 in young men)
→ Mammography/breast MRI
→ Repeat every 3–6 months × 2 years if initially negative
5. EEG — for subclinical seizures in LE / anti-NMDAR
6. EMG/NCS — LEMS (incremental response); PSN (absent SNAPs)
Treatment
| Step | Intervention |
|---|---|
| 1. Treat the tumour | Most critical; eliminates antigenic trigger; most beneficial for LEMS, anti-Ma2, opsoclonus-myoclonus, cell-surface antibody encephalitides |
| 2. First-line immunotherapy | IV methylprednisolone · IVIg · Plasma exchange |
| 3. Second-line immunotherapy | Rituximab · Cyclophosphamide · Mycophenolate · Azathioprine |
| 4. Symptomatic | AEDs (seizures) · 3,4-DAP + pyridostigmine (LEMS) · Diazepam/baclofen (stiff-person) |
Response rule: cell-surface antibody syndromes respond well and are often reversible; intracellular antibody syndromes respond poorly and neuronal loss is irreversible — stabilisation is the realistic goal.
Non-Neurological Paraneoplastic Syndromes
| System | Syndrome | Example Tumour |
|---|---|---|
| Endocrine | SIADH, ectopic ACTH (Cushing's), hypercalcaemia (PTHrP), carcinoid | SCLC, lung, renal |
| Haematological | Trousseau syndrome (DVT/PE), DIC, AIHA | Adenocarcinomas, pancreas |
| Musculoskeletal | Dermatomyositis/polymyositis, hypertrophic osteoarthropathy | Various solid tumours |
| Dermatological | Acanthosis nigricans, paraneoplastic pemphigus, Leser-Trélat sign | GI, lymphoma |
| Renal | Membranous nephropathy, minimal change disease | Solid tumours; Hodgkin |
Recent Development
Immune checkpoint inhibitors (anti-PD-1, anti-CTLA-4) can unmask or de novo trigger paraneoplastic neurological syndromes by amplifying immune responses against shared tumour-neuronal antigens — an increasingly recognised complication in modern oncology. Distinguishing immune-related neurological adverse events from true PNS is an active clinical challenge. — [Farina et al., Lancet Neurol 2024, PMID: 38101905]
Sources: Harrison's Principles of Internal Medicine, 22e · Bradley & Daroff's Neurology in Clinical Practice · Robbins & Cotran Pathologic Basis of Disease · Fishman's Pulmonary Diseases & Disorders · Plum & Posner's Diagnosis and Treatment of Stupor and Coma
This is a shared conversation. Sign in to Orris to start your own chat.