Von willi brand disease
Reading File
Finding Sources
Searching PubMed
"von Willebrand disease"[MeSH Terms]
Reading File
Reading File
Reading File
Searching Images
von Willebrand disease types bleeding
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
I now have comprehensive, well-sourced material for a complete answer. Let me compile it.
Von Willebrand Disease (vWD)
Von Willebrand disease (vWD) is the most common inherited bleeding disorder, with clinically significant bleeding symptoms occurring in approximately 1 in 1,000 individuals. It results from quantitative or qualitative deficiencies in von Willebrand factor (vWF). — Goldman-Cecil Medicine
Pathobiology
VWF is a large multimeric plasma glycoprotein encoded by a 52-exon gene on chromosome 12. It serves two critical roles in hemostasis:
- Platelet adhesion — mediates binding of platelets (via GpIb receptor) to exposed subendothelial collagen at injury sites
- Factor VIII carrier — protects factor VIII from premature proteolytic degradation; the two circulate in a noncovalent complex
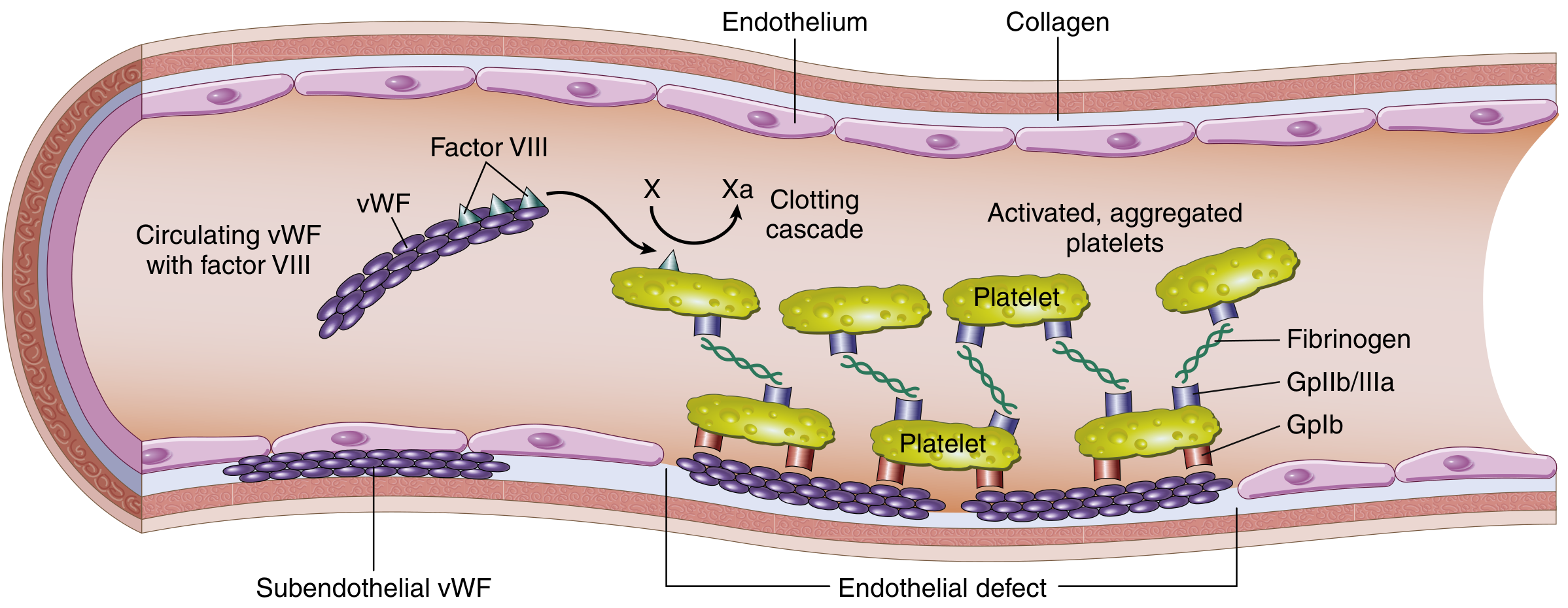
vWF is produced by:
- Endothelial cells (stored in Weibel-Palade bodies, the main plasma source)
- Megakaryocytes (stored in platelet α-granules, ~10–20% of total)
vWF circulates as heterogeneous multimers (2–100 subunits); the high-molecular-weight (HMW) multimers are most functionally active and are cleaved by ADAMTS13 metalloprotease to regulate multimer size. Normal plasma vWF = 50–150 IU/dL. — Goldman-Cecil Medicine
Classification and Types
| Type | Description | vWF Antigen | Activity:Ag Ratio | Multimers | Inheritance |
|---|---|---|---|---|---|
| 1 | Partial quantitative ↓ | ↓ (<30%) | >0.7 | Normal distribution, reduced quantity | Autosomal dominant |
| 2A | Qualitative — loss of HMW multimers (not synthesized) | N to ↓ | <0.7 | HMW absent | Autosomal dominant |
| 2B | Qualitative — "hyperfunctional" HMW multimers → spontaneous platelet aggregation → consumption | N to ↓ | <0.7 | HMW absent; ↑ RIPA at low doses | Autosomal dominant |
| 2M | Qualitative — reduced platelet adhesion, normal multimers | N to ↓ | <0.7 | Normal | Autosomal dominant |
| 2N | Qualitative — reduced binding affinity for Factor VIII only | Normal | >0.7 | Normal | Autosomal recessive |
| 3 | Near-complete absence of vWF | <3 IU/dL | N/A | None | Autosomal recessive |
Type 1 is the classic and most common form (~70–80% of cases). — Robbins & Kumar Basic Pathology
Diagnostic Algorithm
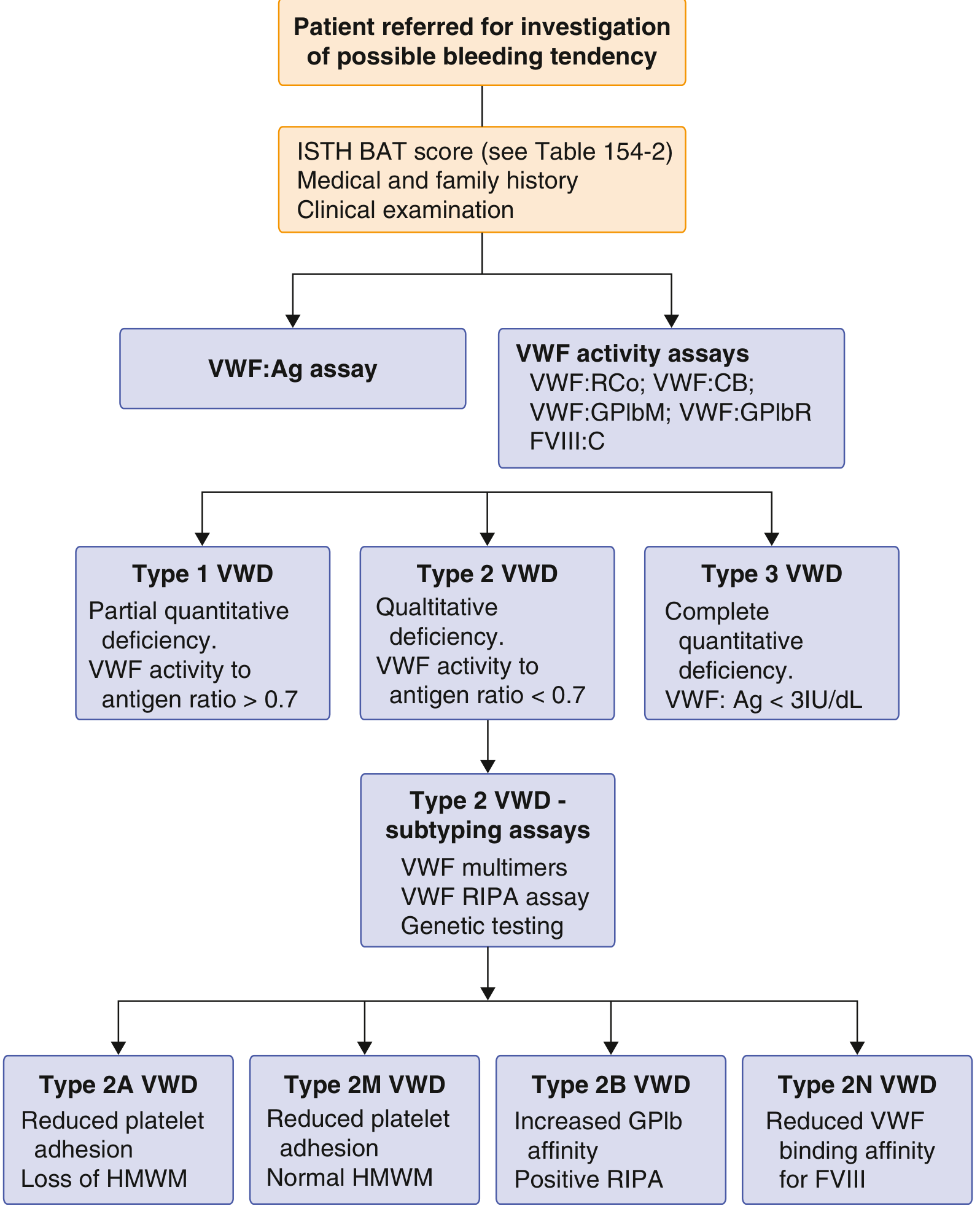
Key Laboratory Tests
- VWF:Ag — measures vWF protein quantity
- VWF activity (ristocetin cofactor, GPIbM, GPIbR assays) — measures functional capacity
- FVIII:C — factor VIII coagulant activity
- VWF multimer analysis — gel electrophoresis to distinguish type 2 subtypes
- RIPA (ristocetin-induced platelet aggregation) — elevated at low doses in type 2B
- PT is usually normal; aPTT may be prolonged in type 3 or severe type 1 (due to low FVIII)
The ISTH Bleeding Assessment Tool (BAT) is used clinically: scores ≥4 in males or ≥6 in females warrant laboratory investigation. — Goldman-Cecil Medicine
Clinical Manifestations
Symptoms correlate with residual vWF levels and subtype:
- Mucocutaneous bleeding: epistaxis, gum bleeding, easy bruising, prolonged wound bleeding
- Heavy menstrual bleeding (menorrhagia): up to 80% of affected women; frequent cause of iron deficiency
- GI bleeding: particularly in types 2 and 3
- Postpartum hemorrhage: both primary and secondary; vWF rises in pregnancy but may be insufficient
- Hemarthrosis: rare, occurs in type 3 (severe FVIII deficiency, resembling hemophilia)
- Clinical disease is more prevalent in females due to greater hemostatic challenges (menstruation, childbirth)
- Type 2B can cause mild thrombocytopenia from platelet consumption
Treatment
| Situation | Treatment |
|---|---|
| Mild–moderate vWD (types 1, some 2) | Desmopressin (DDAVP) 0.3 mcg/kg IV — triggers release of vWF from Weibel-Palade bodies; first-line due to low risk and low cost |
| Adjunctive | Tranexamic acid (antifibrinolytic) — useful for mucosal bleeding, menorrhagia |
| Severe vWD or DDAVP failure | Factor VIII/vWF concentrate (lyophilized) 50 IU/kg — note: factor VIII activity progressively rises 12–40 hours after infusion |
| Emergency/extreme | Fresh frozen plasma (FFP) or cryoprecipitate |
| Type 2B | Avoid DDAVP — releasing more abnormal vWF worsens platelet aggregation/thrombocytopenia |
Desmopressin is contraindicated in type 2B vWD. — Rosen's Emergency Medicine
Key Distinctions from Hemophilia A
| Feature | vWD | Hemophilia A |
|---|---|---|
| Gene/chromosome | VWF / chr 12 | F8 / X chromosome |
| Inheritance | Autosomal dominant (types 1, 2) | X-linked recessive |
| Sex distribution | M = F (clinical bias to F) | Males predominantly |
| Primary defect | vWF ± FVIII | FVIII only |
| Bleeding pattern | Mucocutaneous | Deep (joints, muscles) |
| Hemarthrosis | Rare (type 3 only) | Characteristic |
Sources: Goldman-Cecil Medicine (International Edition); Robbins & Kumar Basic Pathology; Rosen's Emergency Medicine; Tietz Textbook of Laboratory Medicine, 7th Ed.
Disseminate intravascular coagulation
Finding Sources
Searching PubMed
"disseminated intravascular coagulation"[MeSH Terms]
Reading File
Reading File
Searching Images
disseminated intravascular coagulation pathophysiology mechanism
Reading File
Reading File
Loading Image
Loading Image
Excellent — I now have all the material needed. Let me compile the full answer.
Disseminated Intravascular Coagulation (DIC)
DIC is a clinicopathologic syndrome in which simultaneous, uncontrolled activation of both the coagulation and fibrinolytic systems leads to the paradoxical combination of widespread microvascular thrombosis AND diffuse hemorrhage. It is always secondary to an underlying condition — it is never a primary diagnosis. — Henry's Clinical Diagnosis and Management by Laboratory Methods
Pathophysiology
The central event is uncontrolled thrombin generation, which overwhelms physiologic inhibitor systems (antithrombin III, protein C, tissue factor pathway inhibitor [TFPI]) and triggers the cascade below:
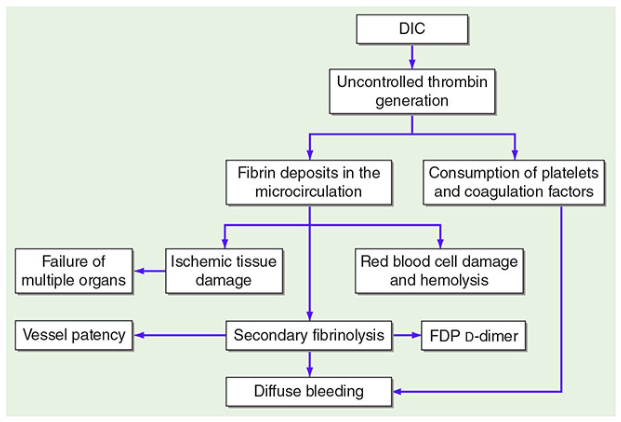
Key sequential events:
- Systemic thrombin generation → fibrin deposition in small vessels of multiple organs
- Consumption of platelets and coagulation factors (especially fibrinogen, factors V, VIII, XIII)
- Activation of plasmin (secondary fibrinolysis) → release of fibrin degradation products (FDPs) and D-dimers
- FDPs further inhibit fibrin polymerization and platelet function → worsening bleeding
- Red blood cell fragmentation on fibrin strands → microangiopathic hemolytic anemia (MAHA)
- Microvascular occlusion → ischemic tissue/organ damage (kidney, liver, brain, adrenals)
— Harrison's Principles of Internal Medicine; Rosen's Emergency Medicine
Causes / Triggers
| Category | Examples |
|---|---|
| Sepsis | Most common cause; gram-negative > gram-positive; fungemia in immunosuppressed |
| Obstetric | Abruptio placentae, amniotic fluid embolism, HELLP syndrome, retained dead fetus (prothrombotic DIC) |
| Malignancy | Acute promyelocytic leukemia (APL) — classic; mucin-secreting adenocarcinomas |
| Massive tissue injury | Trauma, burns, surgery (especially hypothermic circulatory arrest), crush injury |
| Vascular | Giant hemangioma (Kasabach-Merritt), aortic aneurysm |
| Transfusion reaction | ABO incompatibility |
| Snake envenomation | Venom-induced consumption coagulopathy |
Note: Retained dead fetus and APL cause predominantly prothrombotic (non-hemorrhagic) DIC — heparin is appropriate in these settings. — Henry's; Rosen's
Clinical Manifestations
DIC produces a spectrum from compensated (subclinical) to fulminant:
Hemorrhagic features:
- Bleeding from puncture sites, wounds, IV lines, surgical sites (hallmark)
- Purpura, petechiae, ecchymosis
- Mucosal bleeding (GI, urinary, pulmonary)
Thrombotic/ischemic features:
- Purpura fulminans — extensive skin necrosis from dermal microvascular thrombosis
- Acral ischemia, digital gangrene
- Renal failure (oliguria, hematuria)
- CNS dysfunction (altered consciousness, focal deficits)
- Hepatic dysfunction
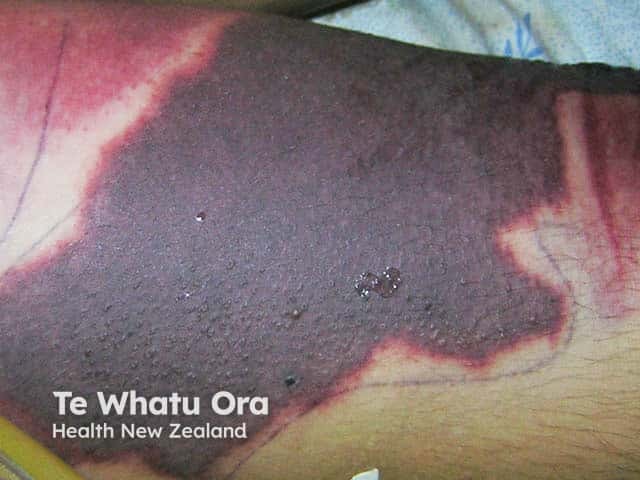
Laboratory Diagnosis
ISTH Scoring System (Overt DIC Score)
Only apply if an underlying condition known to cause DIC is present:
| Test | Result | Points |
|---|---|---|
| Platelet count | >100,000/μL | 0 |
| <100,000/μL | 1 | |
| <50,000/μL | 2 | |
| D-dimer | <0.4 μg/mL | 0 |
| 0.4–4.0 μg/mL | 2 | |
| >4.0 μg/mL | 3 | |
| PT prolongation | <3 sec | 0 |
| 3–6 sec | 1 | |
| >6 sec | 2 | |
| Fibrinogen | >100 mg/dL | 0 |
| <100 mg/dL | 1 |
Score ≥5 = overt DIC (repeat daily). Score <5 = non-overt/early DIC (repeat in 1–2 days). — Henry's Clinical Diagnosis
Full Lab Profile
| Test | Finding in DIC | Mechanism |
|---|---|---|
| Platelet count | ↓ (<100,000/mm³) | Consumed in clotting |
| PT | ↑ Prolonged | Factors II, V consumed |
| aPTT | ↑ Prolonged | Factors II, V, VIII consumed |
| Thrombin time | ↑ Prolonged | Factor II consumed; low fibrinogen |
| Fibrinogen | ↓ Low | Consumption (NB: acute-phase reactant, so may be falsely "normal" early) |
| D-dimer / FDPs | ↑ Elevated | Secondary fibrinolysis |
| Peripheral smear | Schistocytes, helmet cells | RBC fragmentation on fibrin strands |
| Creatinine / UA | May be abnormal | Renal microvascular fibrin deposition |
The absence of schistocytes does not exclude DIC. — Henry's Clinical Diagnosis
DIC vs. Similar Conditions
| Feature | DIC | Severe Liver Disease | Primary Fibrinolysis |
|---|---|---|---|
| Platelets | ↓↓ | ↓ (variable) | Normal |
| PT/aPTT | ↑ | ↑ | ↑ |
| Fibrinogen | ↓↓ | ↓ | ↓↓ |
| D-dimer | ↑↑↑ | ↑ (mild) | ↑ |
| Factor V | ↓ | ↓ (also produced by liver) | Normal/low-normal |
| Factor VIII | ↓ | Normal (not liver-synthesized) | Normal/low-normal |
| Jaundice/splenomegaly | Absent | Present | Absent |
Management
Principle: Treat the underlying cause first
Many episodes are self-limited (e.g., transfusion reactions) or compensated (tumor-associated) and need only supportive care.
Active Bleeding (Hemorrhagic DIC)
| Intervention | Indication |
|---|---|
| Fresh frozen plasma (FFP) | Replenish consumed clotting factors (all factors including V, VIII) |
| Platelet transfusion | Platelets <50,000 or active bleeding |
| Cryoprecipitate | Fibrinogen <100 mg/dL (concentrated fibrinogen, FVIII, vWF, FXIII) |
| Monitor response | ↓ bleeding + ↓ FDPs + ↑ fibrinogen + ↑ platelets → improving; PT normalization lags |
Thrombosis-Predominant DIC
Heparin is considered when fibrin deposition/thrombosis dominates:
- Purpura fulminans
- Retained nonviable fetus (pre-delivery)
- Giant hemangioma (Kasabach-Merritt)
- Acute promyelocytic leukemia (APL)
Heparin is generally not beneficial in meningococcemia, abruptio placentae, severe liver disease, or trauma. LMWH may substitute for UFH.
Agents with No Proven Benefit
- Antithrombin III, PCC, recombinant factor VIIa, activated protein C — none have shown improved outcomes in DIC. — Rosen's Emergency Medicine
Acute vs. Chronic DIC
| Feature | Acute DIC | Chronic DIC |
|---|---|---|
| Onset | Rapid (hours–days) | Slow (weeks–months) |
| Dominant finding | Hemorrhage | Thrombosis |
| Examples | Septic shock, obstetric emergency | Malignancy, dead fetus, giant hemangioma |
| Fibrinogen | Markedly low | Low-normal (compensated synthesis) |
| Lab | Dramatic PT/aPTT prolongation | Subtle abnormalities |
Recent Evidence
A 2025 ISTH systematic review (PMID 40383152) confirmed that sepsis remains the leading etiology of overt DIC globally, with DIC-associated mortality remaining substantially high. A concurrent 2025 systematic review (PMID 40527350) explored platelet function assays as potential supplementary markers to improve DIC diagnosis beyond standard coagulation testing.
Sources: Harrison's Principles of Internal Medicine (21st ed.); Henry's Clinical Diagnosis and Management by Laboratory Methods; Rosen's Emergency Medicine; Goldman-Cecil Medicine (International Edition)
This is a shared conversation. Sign in to Orris to start your own chat.