Ischemic heart disease
Reading File
Finding Sources
Finding Sources
Searching PubMed
"ischemic heart disease" AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have everything needed for a comprehensive, well-illustrated answer. Here is the full response:
Ischemic Heart Disease (IHD)
Definition and Overview
Ischemic heart disease (IHD) is a broad term encompassing several closely related syndromes caused by an imbalance between cardiac blood supply (perfusion) and myocardial oxygen and nutritional demands. It is the leading cause of mortality in the United States and other high-income nations, accounting for approximately 7.5 million deaths worldwide each year.
In more than 90% of cases, IHD results from reduced coronary blood flow secondary to obstructive atherosclerotic vascular disease - making IHD largely synonymous with coronary artery disease (CAD). The remaining cases involve:
- Increased demand (tachycardia, hypertension)
- Diminished blood volume (hypotension, shock)
- Diminished blood oxygenation (pneumonia, CHF)
- Diminished oxygen-carrying capacity (anemia, carbon monoxide poisoning)
- Robbins & Kumar Basic Pathology, p. 351
Epidemiology
- ~800,000 Americans experience an MI each year; roughly half die
- Since peaking in 1963, IHD mortality in the US has declined by 50%
- This decline is attributed to: smoking cessation, hypertension/diabetes treatment, statins, aspirin prophylaxis, thrombolysis, angioplasty/stenting, coronary artery bypass grafting (CABG), and coronary care units
- Remaining challenges: aging baby boomers and the global obesity epidemic
- Robbins & Kumar Basic Pathology, p. 352
Clinical Syndromes
IHD presents as four main clinical syndromes:
| Syndrome | Mechanism |
|---|---|
| Angina pectoris | Ischemia sufficient to cause pain but not myocyte death |
| Myocardial infarction (MI) | Ischemia sufficient to cause cardiomyocyte death |
| Chronic IHD with CHF | Progressive pump failure after MI or cumulative ischemic insults |
| Sudden cardiac death (SCD) | Ischemia-induced lethal ventricular fibrillation |
The term acute coronary syndrome (ACS) applies to the three catastrophic manifestations: unstable angina, MI, and SCD.
Pathogenesis
1. Chronic ("Fixed") Atherosclerotic Occlusion
- <70% occlusion: typically asymptomatic even with exertion
- >70% occlusion ("critical stenosis"): causes symptoms with increased demand → stable angina
- >90% occlusion: may cause symptoms at rest → unstable angina
- Clinically significant plaques occur: first few cm of LAD and LCX, and along the entire RCA
- Gradual occlusion over years allows collateral vessel development, protecting against MI; acute blockage does not allow time for this
2. Acute Plaque Change
In most patients with ACS, the trigger is sudden plaque disruption (rupture or erosion) followed by thrombosis:
- Plaque rupture/fissuring/ulceration exposes thrombogenic contents → rapid thrombosis
- Hemorrhage into the plaque core expands volume, acutely worsening luminal occlusion
- Partial thrombus → subendocardial infarct
- Complete thrombotic occlusion → transmural MI (the typical STEMI)
3. Vasoconstriction / Vasospasm
Triggered by:
- Circulating adrenergic agonists
- Locally released platelet contents
- Imbalance between endothelial relaxing factors (nitric oxide) and contracting factors (endothelin)
- Mediators from perivascular inflammatory cells
- Robbins & Kumar Basic Pathology, pp. 352-354
Types of MI by Location
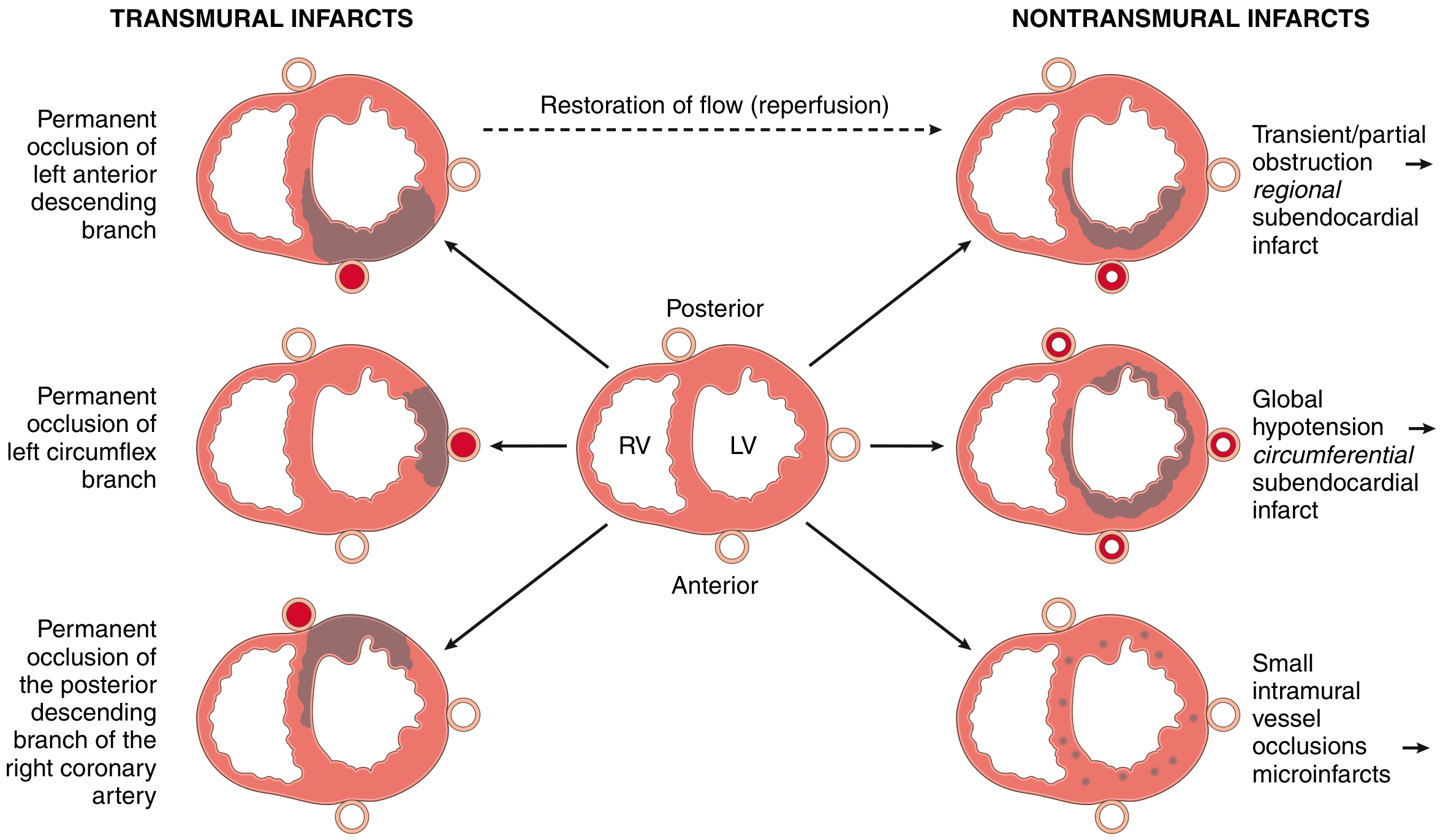
FIG. 9.9: Transmural infarcts (left) result from permanent occlusion of major coronary arteries (LAD, LCX, RCA). Nontransmural infarcts (right) result from transient/partial obstruction, global hypotension, or small vessel occlusions. Reperfusion converts a transmural infarct into a nontransmural (subendocardial) pattern.
- Transmural MI: Involves >50% of ventricular wall thickness; most commonly left ventricle and/or interventricular septum; 15-30% of posterior/posteroseptal MIs extend into RV
- Subendocardial MI: Inner third to half of the ventricular wall; may be circumferential (from global hypotension) or regional (from transient/partial occlusion)
- Microscopic infarcts: Small vessel occlusions from vasculitis, emboli, cocaine-induced vasospasm, or pheochromocytoma
Morphology: Time-Sequence of MI
The gross and microscopic appearance of an MI changes in a highly characteristic sequence:
| Time | Gross Appearance | Microscopy |
|---|---|---|
| 0-12 hrs | Usually not grossly visible; TTC stain shows pale (unstained) area after 3 hrs | Wavy fibers, coagulative necrosis beginning |
| 12-24 hrs | Pale/mottled | Coagulative necrosis + edema, early neutrophilic infiltrate |
| 1-3 days | Pale yellow, well-defined | Dense neutrophilic infiltrate |
| 3-7 days | Hyperemic rim, yellow-tan center | Macrophage infiltration, early phagocytosis |
| 7-10 days | Maximally yellow-tan, soft | Nearly complete macrophage removal of necrotic cells |
| 2-8 weeks | Gray-white scar forming | Granulation tissue (loose connective tissue + capillaries) |
| >2 months | Dense white collagenous scar | Dense fibrous scar, few residual myocytes |
Note: Myocardial necrosis proceeds invariably to scar formation without significant regeneration.
Gross Specimen: Acute MI
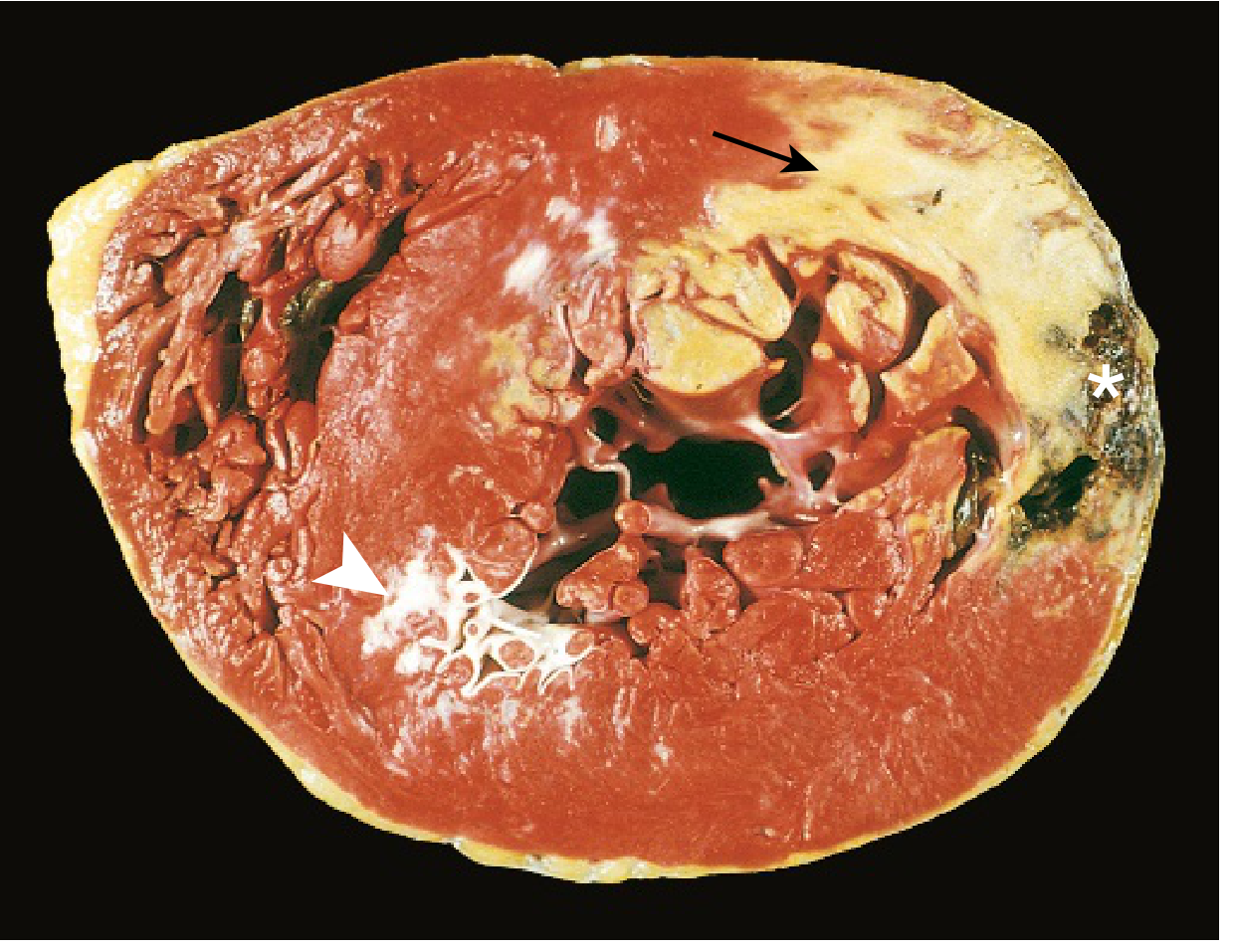
FIG. 9.10: Acute MI of the posterolateral LV. Arrow = pale necrotic area (no TTC staining due to enzyme leakage). Arrowhead = white scar from old remote infarction. Asterisk = ventricular rupture (cause of death).
Microscopic Progression
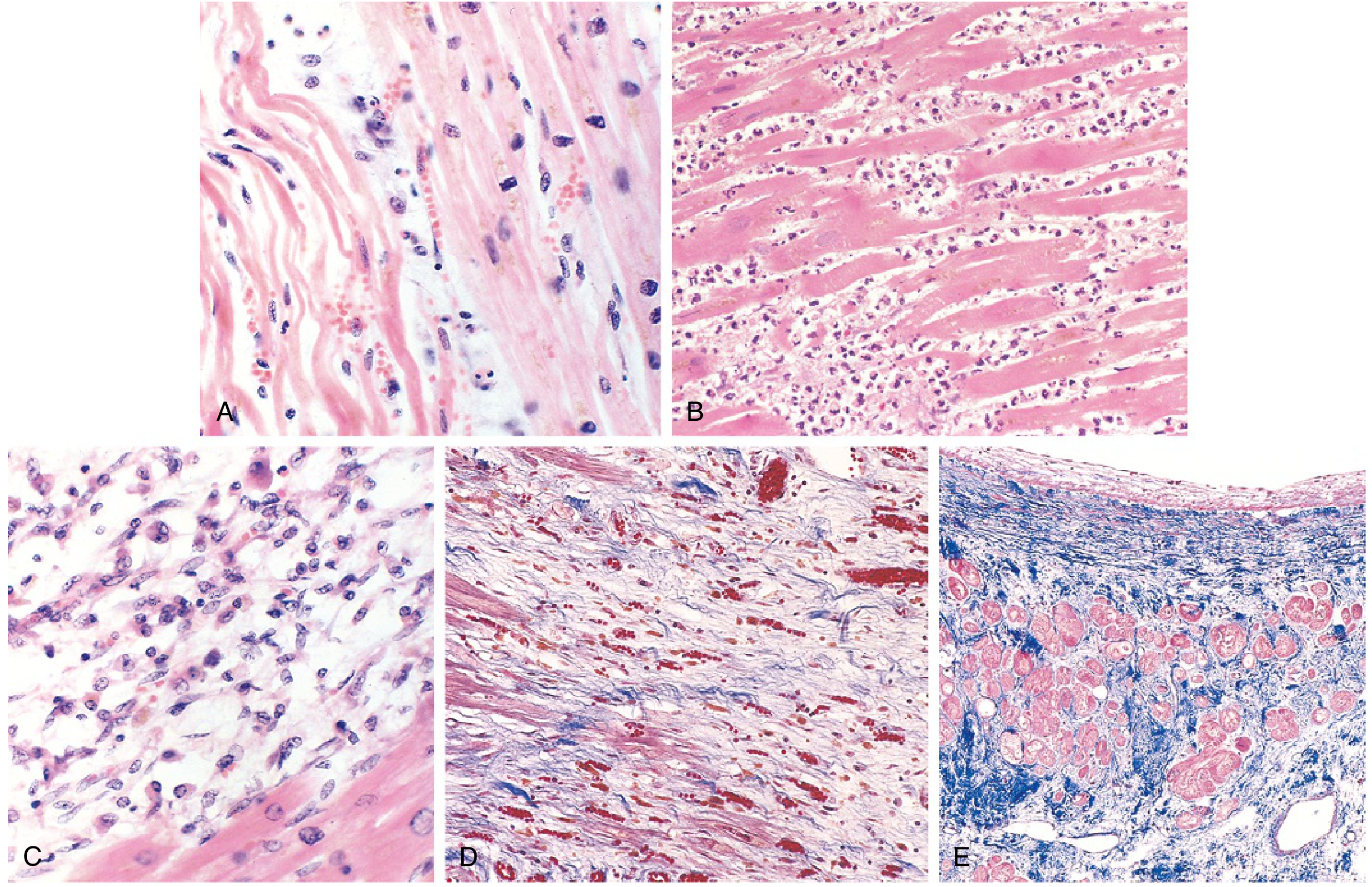
FIG. 9.11: (A) 1-day-old infarct: coagulative necrosis and wavy fibers; (B) 2-3 days: dense neutrophilic infiltrate; (C) 7-10 days: macrophage-mediated phagocytic removal; (D) Granulation tissue with loose connective tissue and capillaries (Masson trichrome - collagen blue); (E) Healed scar: dense collagenous replacement (Masson trichrome).
Reperfusion Injury
When blood flow is restored after ischemia, reperfusion itself causes additional injury through:
- Calcium overload - increased Ca²⁺ influx leads to cytoskeletal contraction, uncontrolled myofibril contraction, and cell death
- Free radical production (O₂⁻, H₂O₂, •OH, peroxynitrite) - damages membrane proteins and phospholipids
- "No-reflow" phenomenon - leukocyte aggregation occludes microvasculature; mediated by phospholipase A₂, prostaglandins, complement activation
- Contraction band necrosis - irreversibly damaged myocytes develop intensely eosinophilic hypercontracted sarcomeres from calcium influx (pathognomonic of reperfused infarcts)
Angina Pectoris - Types
| Type | Mechanism | Features |
|---|---|---|
| Stable (typical) | Fixed stenosis >70%; demand exceeds supply | Predictable with exertion; relieved by rest/nitroglycerin |
| Vasospastic (Prinzmetal) | Coronary artery spasm | Occurs at rest; ECG shows transient ST elevation |
| Unstable | Plaque disruption + partial thrombosis | Occurs with less exertion or at rest; part of ACS; precedes MI |
Clinical Features of MI
- Classic presentation: Severe, crushing substernal chest pain/pressure radiating to neck, jaw, epigastrium, or left arm; lasts minutes to hours; NOT relieved by nitroglycerin or rest
- Silent infarcts: Up to 25% of MIs are entirely asymptomatic, especially in diabetics (autonomic neuropathy) and older adults
- Associated features: Rapid weak pulse, diaphoresis, nausea (especially posterior wall MI), dyspnea (from impaired contractility/mitral valve dysfunction leading to pulmonary congestion)
Diagnosis
- ECG: ST-segment changes, new Q waves
- Cardiac biomarkers: Troponin I/T (most sensitive and specific), CK-MB; rise within hours, peak and fall over days
- Imaging: Echocardiography (wall motion abnormalities), coronary angiography
Complications of MI
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias | Minutes-hours | Most common early complication; VF is the most common cause of sudden death |
| LV failure / cardiogenic shock | Hours-days | Pump failure; 10-15% of hospitalized MIs |
| Myocardial rupture | 3-7 days (peak softening) | Free wall → hemopericardium/tamponade; Septal rupture → VSD; Papillary muscle → acute MR |
| Pericarditis | 2-3 days | Fibrinous/fibrinohemorrhagic; Dressler syndrome (autoimmune) at 2-10 weeks |
| Mural thrombus | Days-weeks | Risk of systemic embolism (stroke) |
| Ventricular aneurysm | Weeks-months | Paradoxical bulging; risk of thrombus, arrhythmia, CHF |
| Progressive CHF | Months-years | Infarct expansion + remodeling |
Chronic IHD
Progressive cardiac decompensation that occurs after acute MI or through accumulated ischemic insults, eventually precipitating mechanical pump failure. Heart failure is typically severe and punctuated by new episodes of angina or infarction. Arrhythmias, CHF, and intercurrent MI account for most associated morbidity and mortality.
Sudden Cardiac Death (SCD)
- Definition: Unexpected death from cessation of normal cardiac electrical activity with hemodynamic collapse
- Incidence: ~450,000 deaths/year in the US
- Underlying cause: 65-70% have coronary atherosclerosis/IHD; ~10% have other causes (hypertrophic cardiomyopathy, channelopathies)
- Mechanism: Usually lethal arrhythmia - ventricular tachycardia, ventricular fibrillation, or asystole
- Successful resuscitation = sudden cardiac arrest (SCA)
- Robbins & Kumar Basic Pathology, p. 368
Management Overview
| Strategy | Examples |
|---|---|
| Risk factor modification | Smoking cessation, statin therapy, BP/diabetes control |
| Antiplatelet therapy | Aspirin, clopidogrel, ticagrelor |
| Anti-ischemic drugs | Beta-blockers, nitrates, calcium channel blockers |
| Reperfusion (acute MI) | Primary PCI (preferred), thrombolysis |
| Revascularization | PCI with stenting, CABG |
| Heart failure management | ACE inhibitors/ARBs, beta-blockers, diuretics, aldosterone antagonists |
| Arrhythmia prevention | ICD for high-risk patients |
Since peaking in 1963, therapy advances have reduced IHD mortality by 50%. The greatest gains came from risk factor intervention (especially smoking cessation, statin use, hypertension treatment) with additional contributions from reperfusion therapies, stenting, and improved acute care.
Sources: Robbins & Kumar Basic Pathology (9780323790185); Braunwald's Heart Disease, 2-Volume Set (9780323722193)
This is a shared conversation. Sign in to Orris to start your own chat.