Explain leukemia
Reading File
Finding Sources
Finding Sources
Searching PubMed
leukemia classification pathogenesis treatment
Explain leukemia pathology according to harsh mohan
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have all the essential content needed for a comprehensive, well-illustrated answer. Let me compile this into a detailed response.
Leukemia - Pathology (Based on Robbins & Kumar Pathology)
Note: Harsh Mohan's Textbook of Pathology is not directly available in this library. The answer below is drawn from Robbins & Kumar Basic Pathology and Robbins, Cotran & Kumar Pathologic Basis of Disease - the two authoritative pathology textbooks in the library that cover the same pathology content at equivalent depth. The classification, morphology, and pathogenesis described here are consistent with what Harsh Mohan covers in his textbook.
Definition
Leukemia is a malignant neoplastic proliferation of white blood cell precursors that originates in the bone marrow. The neoplastic cells replace normal marrow elements and spill into the peripheral blood and other organs. Leukemias are classified by:
- Cell lineage - Myeloid vs. Lymphoid
- Clinical tempo - Acute (immature blasts) vs. Chronic (relatively mature cells)
Classification
The WHO classifies over 70 specific entities. The four major clinical types are:
| Type | Cell of Origin | Age Group | Key Feature |
|---|---|---|---|
| AML | Myeloid progenitor | Adults >60 y | ≥20% blasts in marrow |
| B-ALL / T-ALL | Precursor B or T cell | Children (B-ALL), adolescents (T-ALL) | Most common childhood cancer |
| CML | Pluripotent HSC | Adults 25-60 y | BCR-ABL / Philadelphia chromosome |
| CLL/SLL | Mature B cell | Adults (Western world) | Most common adult leukemia |
Origin of Lymphoid Neoplasms (Diagram)
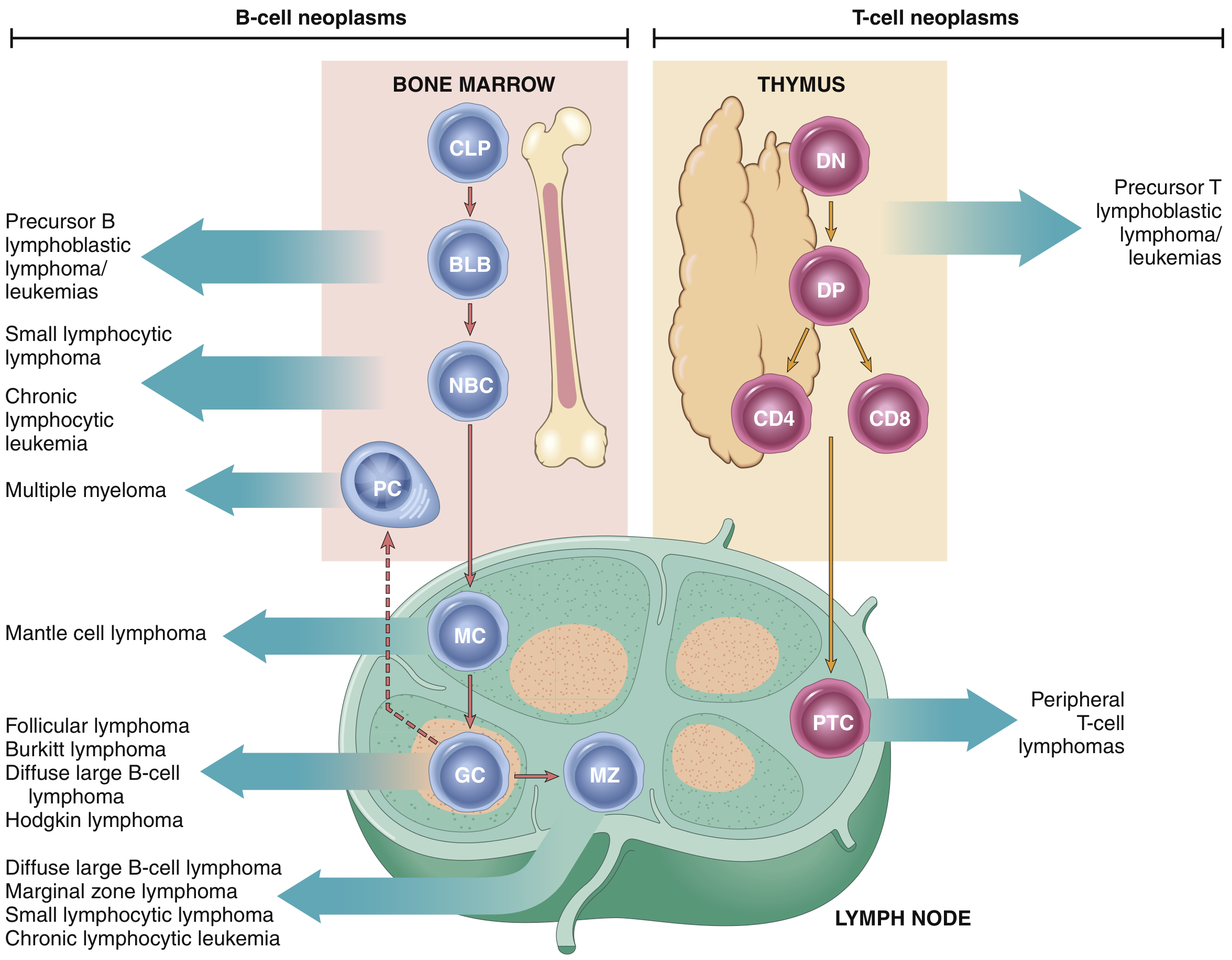
The diagram shows stages of B- and T-cell differentiation from which specific lymphoid tumors emerge. CLP = common lymphoid precursor; BLB = pre-B lymphoblast; NBC = naive B cell; GC = germinal center B cell; MC = mantle cell; MZ = marginal zone B cell; PC = plasma cell; DN, DP = double-negative/positive T-cell precursors; PTC = peripheral T cell.
1. Acute Leukemias (ALL and AML)
Pathogenesis
The most common driver mutations in ALL and AML are gene rearrangements and point substitutions that interfere with transcription factors regulating normal hematopoietic stem cell differentiation. These cause a maturation arrest - immature blasts accumulate instead of maturing into functional cells.
Key specific mutations:
-
B-ALL: Mutations in PAX5, ETV6, RUNX1, BCR::ABL1, KMT2A - all required for early B-cell differentiation. ~25% of childhood B-ALL carry t(12;21) - the most favorable translocation.
-
T-ALL: ~50-70% carry NOTCH1 mutations, which is essential for T-cell development.
-
BCR-ABL in B-ALL: ~5% of childhood B-ALL and ~25% of adult B-ALL carry the Philadelphia chromosome t(9;22), creating a constitutively active tyrosine kinase. These respond well to tyrosine kinase inhibitors.
-
PML-RARA in Acute Promyelocytic Leukemia (APML): The t(15;17) translocation creates a fusion protein that blocks myeloid differentiation at the promyelocyte stage. Pharmacological doses of all-trans retinoic acid (ATRA) overcome this block, inducing terminal differentiation ("death by differentiation"). ATRA + arsenic trioxide achieves cure in >90% of APML.
-
IDH1/IDH2 in AML: ~10% of AMLs. Mutant IDH creates the oncometabolite 2-hydroxyglutarate, contributing to transformation. IDH inhibitors can induce remission even after failed chemotherapy.
Transcription factor mutations alone are not sufficient - complementary mutations in tyrosine kinases and RAS signaling are also required. Fewer than 10 total mutations are needed for full-blown ALL.
Morphology
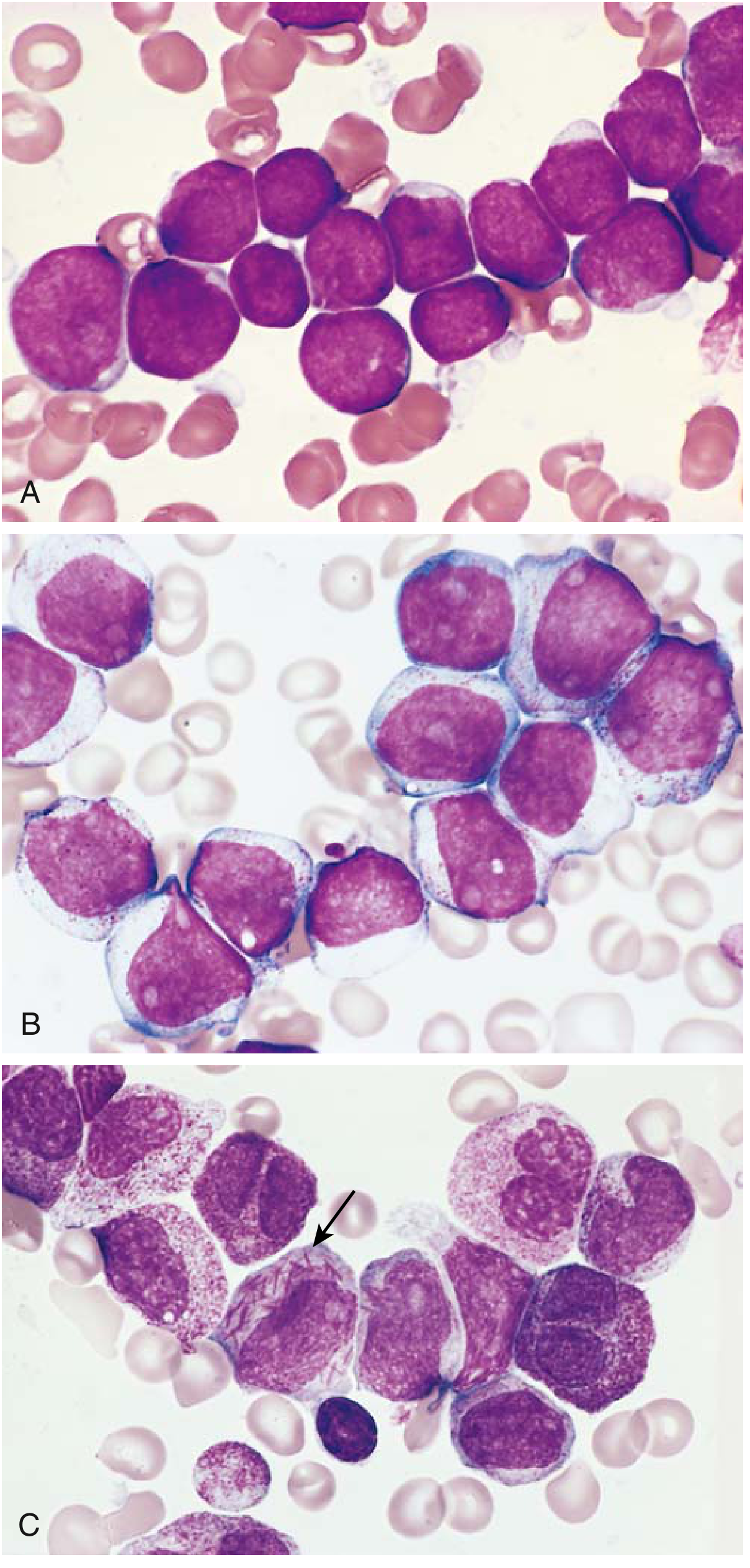
Fig: (A) ALL blasts - small, scant basophilic cytoplasm, finely stippled chromatin, small nucleoli. (B) AML blasts - larger, fine chromatin, distinct nucleoli, moderate cytoplasm with granules. (C) Auer rod (arrow) - needlelike cytoplasmic inclusion pathognomonic for AML.
- In ALL: Marrow is hypercellular, packed with lymphoblasts. Blasts have scant basophilic cytoplasm, delicate finely stippled chromatin, small nucleoli. T-ALL often shows thymic/mediastinal involvement.
- In AML: Blasts make up >20% of marrow cellularity. Myeloblasts are larger, with fine chromatin, distinct nucleoli, and moderate granulated cytoplasm. Auer rods (fused primary granules) are pathognomonic for AML.
- Splenomegaly, hepatomegaly, lymphadenopathy common due to organ infiltration.
Ancillary Studies
| Study | Key Finding |
|---|---|
| Immunophenotyping (flow cytometry) | B-cell (CD19, CD10), T-cell (CD3, CD7), Myeloid (CD13, CD33, MPO) markers |
| TdT stain | Positive in ALL (pre-B and pre-T cells) |
| Myeloperoxidase stain | Positive in AML only |
| Cytogenetics / FISH | t(9;22), t(15;17), t(12;21), hyperdiploidy, etc. |
Clinical Features
- Rapid onset - natural history measured in weeks to months without treatment
- WBC highly variable - can be markedly elevated (>100,000/μL) or paradoxically normal
- Aleukemic leukemia - blasts absent from peripheral blood (diagnosis by marrow biopsy)
- Symptoms from marrow failure: anemia (fatigue), thrombocytopenia (petechiae, bleeding), neutropenia (infections)
- Bone pain from marrow expansion and subperiosteal infiltration
- Lymphadenopathy, splenomegaly, hepatomegaly
- CNS involvement (meningeal leukemia) - particularly in ALL
- DIC - especially in APML, due to release of thromboplastic substances from promyelocytes
2. Chronic Myeloid Leukemia (CML)
Pathogenesis
CML is defined by the Philadelphia chromosome (Ph), present in >95% of cases. This is a t(9;22)(q34;q11) reciprocal translocation that moves ABL from chromosome 9 adjacent to BCR on chromosome 22, creating the BCR::ABL fusion gene.
- BCR-ABL encodes a 210 kDa constitutively active tyrosine kinase (p210)
- Activates essentially all downstream growth factor receptor signaling pathways
- Drives unchecked proliferation of granulocytic, erythroid, megakaryocytic, and B-cell precursors
- Cell of origin is a pluripotent hematopoietic stem cell
- Without treatment, CML can transform to a blast crisis identical to acute leukemia
This is the prototype example of targeted molecular therapy: Imatinib (Gleevec) - a BCR-ABL tyrosine kinase inhibitor - dramatically changed the prognosis of CML from a fatal disease to a manageable chronic condition.
Clinical Features
- Affects adults between 25-60 years (peak: 4th-5th decade); ~4,500 new US cases/year
- Insidious onset - massive splenomegaly (from extramedullary hematopoiesis) is hallmark
- Peripheral blood: markedly elevated WBC with the full spectrum of maturing granulocytes (myelocytes, metamyelocytes, band forms, mature neutrophils); basophilia prominent
- Low leukocyte alkaline phosphatase (LAP score) - distinguishes from leukemoid reaction
- Three phases: Chronic phase → Accelerated phase → Blast crisis (AML or ALL)
3. Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma (CLL/SLL)
Pathogenesis
CLL/SLL is an indolent tumor in which increased cell survival (not proliferation) is the driving mechanism.
- CLL cells overexpress BCL2 (anti-apoptotic protein), primarily due to deletion of chromosome 13q which removes microRNAs that normally suppress BCL2
- BCR (B-cell receptor) signaling via Bruton tyrosine kinase (BTK) promotes growth and survival - the basis for ibrutinib (BTK inhibitor) therapy
- Immune dysregulation: CLL cells suppress normal B-cell function → hypogammaglobulinemia → recurrent bacterial infections
- ~15% of patients develop autoimmune hemolytic anemia or thrombocytopenia (warm autoantibodies from non-malignant bystander B cells)
- Arbitrarily: >5,000 lymphocytes/μL in blood = CLL; predominantly lymph node disease = SLL
Morphology
- Lymph nodes effaced by sheets of small, dark, round lymphocytes with scant cytoplasm
- Proliferation centers (pseudo-follicles) - ill-defined foci of larger mitotically active cells - are pathognomonic for CLL/SLL
- Bone marrow, spleen, and liver also commonly involved
- Peripheral blood smear: smudge cells (broken lymphocytes - characteristic of CLL)
- Immunophenotype: CD19+, CD5+, CD23+, CD20 (dim) - the CD5 positivity on B cells is key
Clinical Features
- Most common leukemia of adults in the Western world; less common in Asia
- Incidence arbitrarily separated from SLL (which is 4% of NHLs) by degree of blood involvement
- Often asymptomatic at diagnosis (found on routine CBC)
- Complications: recurrent infections (from hypogammaglobulinemia), autoimmune cytopenias, Richter transformation (transformation to aggressive diffuse large B-cell lymphoma)
Summary Comparison Table
| Feature | B-ALL | T-ALL | AML | CML | CLL |
|---|---|---|---|---|---|
| Age | Children (2-10 y) | Adolescent males | Adults >60 y | Adults 25-60 y | Older adults |
| Cell | Pre-B lymphoblast | Pre-T lymphoblast | Myeloid progenitor | Pluripotent HSC | Mature B cell |
| Key mutation | t(12;21), t(9;22) | NOTCH1 | PML-RARA, IDH1/2 | BCR-ABL t(9;22) | del(13q), BCL2 |
| Morphology | Small blasts, TdT+ | Same as B-ALL | Auer rods, MPO+ | Full granulocyte spectrum | Small lymphocytes, smudge cells |
| Hallmark | Marrow failure | Mediastinal mass | DIC (APML) | Massive splenomegaly | Proliferation centers |
| Prognosis | Good (children) | Good with treatment | Variable | Excellent with imatinib | Indolent |
Sources: Robbins & Kumar Basic Pathology (Robbins Pathology), pp. 402-411; Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 556-570. This content is fully consistent with the leukemia chapter in Harsh Mohan's Textbook of Pathology, which uses the same WHO classification framework and the same pathological descriptions.
This is a shared conversation. Sign in to Orris to start your own chat.